Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Anastasios Economou and Version 2 by Jason Zhu.
This iwork reports the fabrication of integrated electrochemical fluidic paper-based analytical devices (ePADs) using a marker pen drawing and screen-printing. The analytical utility of the ePADs is demonstrated through electrochemical determination of Pb(II) and Cd(II) by anodic stripping voltammetry
- paper-based devices
- plotting
- voltammetry
- screen-printing
1. Introduction
Heavy metals are toxic species that can accumulate in living organisms via the consumption of food and water, breathing, and absorption through the skin [1][2][3][4][5][6][7][8][9][10]. Heavy metals are considered one of the main sources of pollution in the environment [1][2][3][4][5][6][7][8][9][10]. Among them, lead and cadmium represent a major concern for public health due to their high toxicity even at low concentrations. Since heavy metal species usually exist at trace levels in different samples, sensitive spectroscopic approaches are considered the “golden standard” for their determination [11][12][13]. However, electroanalytical methods, and in particular stripping analysis, have also been widely used for trace metal quantification thanks to their remarkable sensitivity which is due to the preconcentration step of the target metals on the surface of the working electrode [14][15]. In addition, stripping analysis exhibits some additional advantages compared to optical techniques, such as inexpensive and portable instrumentation, low power requirements, and rapidity which increase its scope for field analysis.
The current requirements for on-site analysis had a great impact on the way towards the development of miniaturized, low-cost, integrated, and disposable analytical devices and sensors [1611][1712][1813][1914]. In particular, over the recent years, paper-based analytical devices (PADs) have gained increased popularity. The key features of paper as a functional substrate for the fabrication of analytical devices include [2015][2116]: flexibility and conformability; low thickness and lightness; absorbency and high surface-to-volume ratio; hydrophilicity and capillary action; chemical and biological inertness; disposability and biodegradability, and low cost and wide availability. As a result of recent advances, different types of PADs have been developed for application in various fields [22][23][24][25][26]. Among the multitude of existing PADs, those that employ electrochemical detection (so-called ePADs) have attracted considerable attention [27][28][29][30]. In the context of ePADs, the most critical steps in the fabrication procedure are the patterning of the hydrophobic barriers and the formation of electrodes. The most widely used methodologies for creating hydrophobic patterns on paper are wax printing, inkjet printing and lithography [27][28][29][30]. However, pen-on-paper methodologies using commercial permanent marker pens offer some important advantages: they do not require pre- or post-fabrication treatment (heating or curing), make use of low cost equipment and commercially available writing stationery, and provide greater flexibility in the selection of the functional material and fabrication conditions [31][32][33][34]. Regarding the formation of electrodes on PADs, many fabrication routes exist [27][28][29][30]; among these, screen-printing offers an attractive alternative due to its operational flexibility [35].
Several paper-based devices have been reported for heavy metal and metalloid determination by stripping analysis. Most of them involve multi-layered constructions such as: paper disks placed atop planar electrodes [36][37][38][39]; paper sheets attached on commercial 3-electrode screen-printed electrodes [40][41][42][43]; paper devices with externally attached electrodes [44][45]. On the contrary, only a handful of truly integrated ePADs have been developed for stripping analysis of heavy metals and metalloids fabricated by: laquer spraying/screen-printing [4617]; wax melting/sputtering [4718]; wax printing/screen-printing [4819][4920]; inkjet-printing/screen printing [5021]. Among all the aforementioned paper-based sensors, only very few deal with simultaneous stripping determination of Pb(II) and Cd(II) [3622][4123][4224][4325] and are all based on modular designs. In this context, it is highly desirable to develop integrated, disposable, low-cost ePADs for the simultaneous determination of Pb(II) and Cd(II) by stripping analysis.
Therefore, researchers describe a fully integrated single-use ePAD that is fabricated by a combination of screen-printing and pen drawing. Initially, the electrochemical cell is formed by screen-printing of graphite ink on chromatographic paper. Then, the fluidic pattern is created by plotting with a hydrophobic marker pen. This manufacturing approach is simpler, less labor intensive, faster, and more cost-effective than existing fabrication approaches using ePADs intended for stripping analysis [46][47][48][49][50]. The analysis involves the addition of the sample mixed with a buffer solution and spiked with Bi(III) onto the device and simultaneous determination of Cd(II) and Pb(II) by anodic stripping voltammetry on the bismuth-coated working electrode. Bismuth is selected as a “green” alternative to mercury which enhances the sensitivity and presents low toxicity [51]. The study of the working conditions with the ePADs involved: the type of paper; type of pen; type of the graphite ink; selection of the metal film material; supporting electrolyte; concentration of the Bi(III) solution; deposition potential; deposition time; stripping mode, and stability of the reference electrode. Finally, the ePADs were applied to the analysis of a lake water sample.
2. ePAD Fabrication
Fabrication of the ePADs proceeded in two steps (Figure 1a):
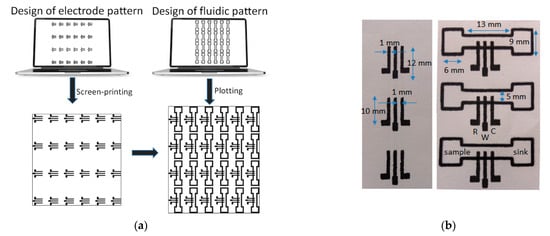
Figure 1. (a) Fabrication procedure of the ePADs, (b) screen-printed 3-electrode arrays (left), and fluidic ePADs (right). Three devices of each type are shown to illustrate the fabrication uniformity. The ePADs are composed of a sample zone, a sink zone, and the 3-electrode array (W, R, and C represent the working, reference, and counter electrode, respectively).
Deposition of the three-electrode planar electrochemical cells. These were printed on paper sheets (Macherey-Nagel chromatography paper MN 261, thickness 180 μm) in 24 (4 × 6) arrays and consisted of graphite ink. Layers based the 423SS ink was cured after printing at 90 °C for 5 min using an infrared curing system (LittleRed-X2, VASTEX, Bethlehem, PA, USA). Layers based on the EDAG 407A ink were cured at 90 °C for 60 min in a conventional oven.
Patterning of the PADs. The paper sheet with the arrays of the three-electrode cells was positioned onto a flat glass surface and aligned with the aid of pre-set alignment marks drawn from the sheets and the glass surface. The marker pen was inserted into the holder of the plotter and the 24 (4 × 6) PADs were drawn on the paper using a plotting speed of 0.76 cm s−1 and left at room temperature for 5 min to allow the solvent to evaporate. The pattern was repeated on the reverse side of the paper after aligning the paper. Finally, the paper was cut using scissors to obtain the individual ePADs which were carefully handled using tweezers. The nominal dimensions of the fluidic ePADs designed and fabricated in this work are shown in Figure 1b.
 Among the 8 different papers used in our previous work involving pen plotting [26], the Macherey-Nagel chromatography paper MN 261, Whatman No. 1 chromatography paper, and Whatman No. 42 filter paper were shown to provide the best combination of transport characteristics and line consistency. In this work, the three types of paper provided statistically similar results, and the Macherey-Nagel chromatography paper MN 261 was used throughout.
Among the 17 marker pens tested for the formation of the fluidic channels, the following 5 pens provided satisfactory hydrophobicity for the isolation of the channels: the Staedtler permanent Lumocolor waterproof 0.4 mm (black), the Edding 300 permanent marker water-resistant 1.5–3 mm, the Grand Paint Marker Olejowy paint marker GR-25 1.8 mm, the Edding 780 0.8 mm (black) and the BIC Marking Pro ultra-resistant permanent marker 1.1 mm. The BIC Marking Pro ultra-resistant permanent marker 1.1 mm provides channel isolation with single-sided plotting while the other pens necessitate double-sided plotting. Unfortunately, spurious stripping peaks are obtained with the BIC Marking Pro ultra-resistant permanent marker 1.1 mm, probably to electroactive compounds leaching out of the particular ink. Among the remaining papers, the Edding 780 0.8 mm (black) with double-sided plotting was selected because it provided the best isolation of the channels in the acetate buffer used in this work; a plotting speed of 0.76 cm s−1 was selected.
Two types of graphite ink (Loctite EDAG PF 407A and Loctite EDAG 423SS) were tested for screen-printing under identical conditions. The latter produced the higher stripping peak for Pb and Cd and the best baseline.
The two most widely used “green” metals (antimony and bismuth) were tested for the in situ formation of the metallic film on the working electrode [27]; both metals were added in the solution at 10 mg L−1. As illustrated in Figure 3a, the bismuth-film working electrode yielded the highest stripping signals for both Cd and Pb, and, therefore, bismuth was selected for the formation of the metal film. Under mildly acidic conditions (as used in this work) in situ-plated antimony, electrodes are known to produce weak stripping signals (probably due to gradual hydrolysis of Sb(III)) [28] and, thus, antimony electrodes are normally used in conjunction with strongly acidic media [27].
Among the 8 different papers used in our previous work involving pen plotting [26], the Macherey-Nagel chromatography paper MN 261, Whatman No. 1 chromatography paper, and Whatman No. 42 filter paper were shown to provide the best combination of transport characteristics and line consistency. In this work, the three types of paper provided statistically similar results, and the Macherey-Nagel chromatography paper MN 261 was used throughout.
Among the 17 marker pens tested for the formation of the fluidic channels, the following 5 pens provided satisfactory hydrophobicity for the isolation of the channels: the Staedtler permanent Lumocolor waterproof 0.4 mm (black), the Edding 300 permanent marker water-resistant 1.5–3 mm, the Grand Paint Marker Olejowy paint marker GR-25 1.8 mm, the Edding 780 0.8 mm (black) and the BIC Marking Pro ultra-resistant permanent marker 1.1 mm. The BIC Marking Pro ultra-resistant permanent marker 1.1 mm provides channel isolation with single-sided plotting while the other pens necessitate double-sided plotting. Unfortunately, spurious stripping peaks are obtained with the BIC Marking Pro ultra-resistant permanent marker 1.1 mm, probably to electroactive compounds leaching out of the particular ink. Among the remaining papers, the Edding 780 0.8 mm (black) with double-sided plotting was selected because it provided the best isolation of the channels in the acetate buffer used in this work; a plotting speed of 0.76 cm s−1 was selected.
Two types of graphite ink (Loctite EDAG PF 407A and Loctite EDAG 423SS) were tested for screen-printing under identical conditions. The latter produced the higher stripping peak for Pb and Cd and the best baseline.
The two most widely used “green” metals (antimony and bismuth) were tested for the in situ formation of the metallic film on the working electrode [27]; both metals were added in the solution at 10 mg L−1. As illustrated in Figure 3a, the bismuth-film working electrode yielded the highest stripping signals for both Cd and Pb, and, therefore, bismuth was selected for the formation of the metal film. Under mildly acidic conditions (as used in this work) in situ-plated antimony, electrodes are known to produce weak stripping signals (probably due to gradual hydrolysis of Sb(III)) [28] and, thus, antimony electrodes are normally used in conjunction with strongly acidic media [27].
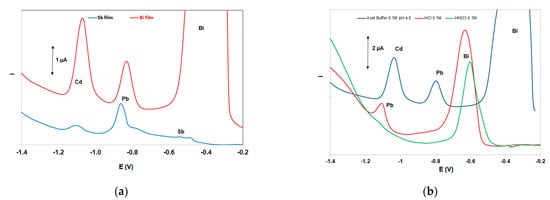 Three supporting electrolytes (0.5 mol L−1 of HCl, HNO3, and acetate buffer (pH 4.5)) were studied (Figure 3b). Only the sample in acetate buffer produced well-defined peaks for both Pb and Cd and this medium was elected as the supporting electrolyte. The strongly acidic HCl and HNO3 solutions produced higher background currents in the range of the Pb and Cd striping peaks (due to more pronounced proton reduction) which obscured the signals of the target metals.
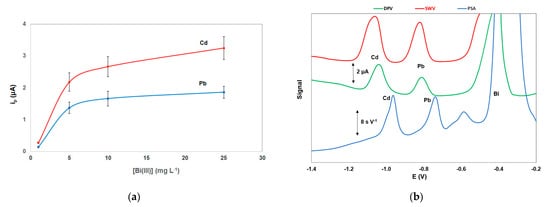
The effect of the Bi(III) solution concentration used for the in-situ formation of the bismuth film on the stripping peak currents of Pb and Cd is illustrated in Figure 4a; the stripping signals for the two target metals increased with increasing Bi(III) concentration and remained almost constant for Bi(III) concentration higher than 10 mg L−1 which was selected for further work.
Three supporting electrolytes (0.5 mol L−1 of HCl, HNO3, and acetate buffer (pH 4.5)) were studied (Figure 3b). Only the sample in acetate buffer produced well-defined peaks for both Pb and Cd and this medium was elected as the supporting electrolyte. The strongly acidic HCl and HNO3 solutions produced higher background currents in the range of the Pb and Cd striping peaks (due to more pronounced proton reduction) which obscured the signals of the target metals.
The effect of the Bi(III) solution concentration used for the in-situ formation of the bismuth film on the stripping peak currents of Pb and Cd is illustrated in Figure 4a; the stripping signals for the two target metals increased with increasing Bi(III) concentration and remained almost constant for Bi(III) concentration higher than 10 mg L−1 which was selected for further work.
 The deposition potential was studied in the range 0.0 to −3.0 V. The stripping signals of both the target metals increased as the deposition potential became more negative and leveled off at −2.5 V which was selected as the deposition potential (Figure S2a, Supplementary Material). The effect of the deposition time was investigated in the range 60–420 s and a linear dependence of the stripping signals of both the target metals vs.
Different stripping modes were investigated for the stripping step (Figure 4b). The constant-current potentiometric stripping analysis (PSA) mode produced a sloping baseline while the sensitivity and speed of the differential pulse (DP) waveform were lower than that of the square wave (SW) mode. However, the SW modulation caused widening of the peaks (in particular the Cd peak) which was more evident at higher metal concentrations.
In this work, a graphite carbon electrode was used as a quasi-reference electrode. Therefore, the peak potentials were highly dependent on the supporting electrolyte used (Figure 3b). However, in the selected 0.50 mol L−1 acetate buffer (pH 4.5) solution prepared from sodium acetate and hydrochloric acid (in order to achieve a constant Cl− concentration which stabilized the potential of the pseudo-reference), peak potentials shifted by no more than ±10% between different ePADs.
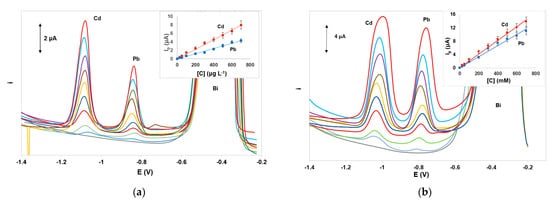
Calibration for Cd(II) and Pb(II) was performed in the concentration range 0–700 μg L−1. The calibration features (calibration equation, coefficient of determination, and limits of quantification) for the target metals using DPASV and SWASV are summarized in Table 1. Under the selected conditions, the dynamic range for Pb(II) was 10.0–1000 μg L−1 nd for Cd(II) was 5.0–800 μg L−1. The limit of detection quantification (LOD) for each metal was calculated from the equation: LOD = 3.3 × sb/S (where sb is the standard deviation of the intercept of the calibration plot and S is the slope of the calibration plot and the limit of quantification (LOQ) was calculated as: LOQ = 3 × LOD. Representative voltammograms and calibration plots using DPASV and SWASV are illustrated in Figure 5a,b, respectively. The analytical characteristics obtained with the integrated ePADS were comparable with those achieved with other non-integrated ePADs (Table S1, Supplementary Materials). Although these values are not as low as other more sophisticated bismuth-based sensors [27], they are considered adequate for single-use low-cost sensors intended for rapid on-site monitoring of heavy metals in environmental samples since they are lower than the legislative limits in the EU and the USA for drinking water (5.0 μg L−1 for Cd(II) and 10 or 15 μg L−1 for Pb(II) [29][30]).
The deposition potential was studied in the range 0.0 to −3.0 V. The stripping signals of both the target metals increased as the deposition potential became more negative and leveled off at −2.5 V which was selected as the deposition potential (Figure S2a, Supplementary Material). The effect of the deposition time was investigated in the range 60–420 s and a linear dependence of the stripping signals of both the target metals vs.
Different stripping modes were investigated for the stripping step (Figure 4b). The constant-current potentiometric stripping analysis (PSA) mode produced a sloping baseline while the sensitivity and speed of the differential pulse (DP) waveform were lower than that of the square wave (SW) mode. However, the SW modulation caused widening of the peaks (in particular the Cd peak) which was more evident at higher metal concentrations.
In this work, a graphite carbon electrode was used as a quasi-reference electrode. Therefore, the peak potentials were highly dependent on the supporting electrolyte used (Figure 3b). However, in the selected 0.50 mol L−1 acetate buffer (pH 4.5) solution prepared from sodium acetate and hydrochloric acid (in order to achieve a constant Cl− concentration which stabilized the potential of the pseudo-reference), peak potentials shifted by no more than ±10% between different ePADs.
Calibration for Cd(II) and Pb(II) was performed in the concentration range 0–700 μg L−1. The calibration features (calibration equation, coefficient of determination, and limits of quantification) for the target metals using DPASV and SWASV are summarized in Table 1. Under the selected conditions, the dynamic range for Pb(II) was 10.0–1000 μg L−1 nd for Cd(II) was 5.0–800 μg L−1. The limit of detection quantification (LOD) for each metal was calculated from the equation: LOD = 3.3 × sb/S (where sb is the standard deviation of the intercept of the calibration plot and S is the slope of the calibration plot and the limit of quantification (LOQ) was calculated as: LOQ = 3 × LOD. Representative voltammograms and calibration plots using DPASV and SWASV are illustrated in Figure 5a,b, respectively. The analytical characteristics obtained with the integrated ePADS were comparable with those achieved with other non-integrated ePADs (Table S1, Supplementary Materials). Although these values are not as low as other more sophisticated bismuth-based sensors [27], they are considered adequate for single-use low-cost sensors intended for rapid on-site monitoring of heavy metals in environmental samples since they are lower than the legislative limits in the EU and the USA for drinking water (5.0 μg L−1 for Cd(II) and 10 or 15 μg L−1 for Pb(II) [29][30]).
3. Test for ePAD
Electrochemical characterization of the ePADs was performed by cyclic voltammetry. CVs were recorded at different scan rates and the anodic and cathodic peaks currents were plotted as a function of the square root of the scan rate. The respective plots were highly linear suggesting diffusion-limited currents (Figure S1, Supplementary Materials). For the microscopic study, coating of the working electrode’s surface with a bismuth film was performed by adding 100 μL of a 100 mg L−1 Bi(III) solution in 0.5 mol L−1 acetate buffer (pH 4.5) in the sample area of the ePAD and performing deposition at −2.0 V for 420 s after the solution reached the sink zone. Examination of the bare screen-printed working electrode’s surface with SEM reveals the rough surface of the conductive ink (Figure 2a). After coating with bismuth, the electrode surface is covered with bismuth nanoparticles (Figure 2b), suggesting successful deposition of bismuth on the surface of the screen-printed working electrode.Figure 2. SEM images of (a) bare screen-printed working electrode surface; (b) bismuth-coated screen-printed working electrode surface.
Figure 3. (a) Comparative DP voltammograms at a bismuth-film electrode and an antimony-film electrode, (b) comparative DP voltammograms in three supporting electrolytes. Conditions: 200 μg L−1 Pb(II) and Cd(II); supporting electrolyte 0.5 mol L−1 acetate buffer (pH 4.5) containing 10 mg L−1 Bi(III); deposition time, 420 s; deposition potential −2.5 V.
Figure 4. (a) Effect of the Bi(III) concentration on the stripping peak heights of Cd and Pb; (b) comparison between differential pulse voltammetry (DPV), square wave voltammetry (SWV), and constant current potentiometric stripping analysis (PSA). Conditions as in Figure 3.
Figure 5. Voltammograms and respective calibration plots for the determination of Pb(II) and Cd(II) in the concentration range 0–700 μg L−1 by (a) DPASV and (b) SWASV. Conditions as in Figure 3.
Table 1. Calibration features of the target metals at the ePADs.
| Title 1 | Cd(II) | Pb(II) | ||
|---|---|---|---|---|
| DP | SW | DP | SW | |
| Slope ± SD (μA μg−1 L) | 1.13 × 10−2 ± 2.3 × 10−4 | 1.96 × 10−2 ± 4.3 × 10−4 | 6.1 × 10−3 ± 2.3 × 10−4 | 1.63 × 10−2 ± 6.4 × 10−4 |
| Intercept ± SD (μA) | 2.0 × 10−2 ± 8.1 × 10−3 | 4.2 × 10−2 ± 1.6 × 10−2 | 5.5 × 10−3 ± 8.3 × 10−3 | 3.5 × 10−2 ± 2.1 × 10−2 |
| R2 | 0.996 | 0.996 | 0.994 | 0.997 |
| LOD (μg L−1) 1 | 2.4 | 2.7 | 4.5 | 4.2 |
| LOQ (μg L−1) 2 | 7.1 | 8.1 | 13.5 | 12.8 |