Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 1 by Charles Joseph.
New approaches are required to successfully intervene therapeutically in neurodegenerative diseases. Addressing the earliest phases of disease, blood brain barrier (BBB) leak before the accumulation of misfolded proteins has significant potential for success.
- glymphatic flow
- blood brain barrier
- Alzheimer disease
- neurodegenerative diseases
1. Introduction
In the wake of disappointing treatment trials of Alzheimer modifying drugs, a fresh therapeutic approach to this and other neurodegenerative diseases is necessary. Approaching the initial disease phase, the pathological leak of the blood brain barrier and consequent obstruction of glymphatic flow is worthy of therapeutic investigation. To do so requires identification of these very early pathophysiologic changes developing before the accumulation of misfolded proteins and significant cognitive decline has developed. [1,2][1][2]. Looking at sporadic AD in stages, the initial insult is the blood brain barrier (BBB) leak, triggered by endothelial and pericyte damage resulting from circulating low level upregulated pro-inflammatory factors via several potential sources [1,3][1][3]. Senescence, morbid obesity, chronic infection, autoimmune disease, diabetes mellitus, hypertension and head injury are the most common factors [3,4,5][3][4][5]. Similar to a computer hacker, the attacks are relentless over one’s lifetime and once the defenses are overwhelmed, either because of sheer volume of pro-inflammatory factors or reduced effectiveness of the CNS immune response (e.g., microglia and astrocyte responses), the BBB is breached [6,7][6][7]. The circulating inflammatory factors cause endothelial cell up regulation of Matrix Metallo Protease Enzyme-9 (MMPE-9), expressed on the cell surface triggering release of reactive oxygen species ROS from circulating and inherent macrophages and immune cells. Damage to the tight junctions, endothelium and pericytes results [8,9][8][9]. The consequence is a leak into the interstitium of normally excluded substances, so called damage associated molecular patterns (DAMPS) and pathogen associated molecular patterns (PAMPS) which alter the normal metabolic machinery resulting in distorted protein synthesis and degradation [10,11,12][10][11][12]. Additionally, both misfolded proteins and toxins overwhelm the innate immune cells (microglia and macrophages) normally clearing them, either by degradation or by outflow from the venous or glymphatic systems (Figure 1a,b) [13,14,15,16][13][14][15][16]. As a consequence of the pericyte damage that normally anchors the aquaporin 4 water channels to astrocyte end feet via expressed laminin and dystroglycan proteins, the glymphatic drainage system effectively shuts down as the untethered AQ 4 channels return to the astrocyte soma. The glymphatic system is a major pathway of metabolite clearance from the brain parenchyma (Figure 1 and Figure 2) [17,18][17][18].
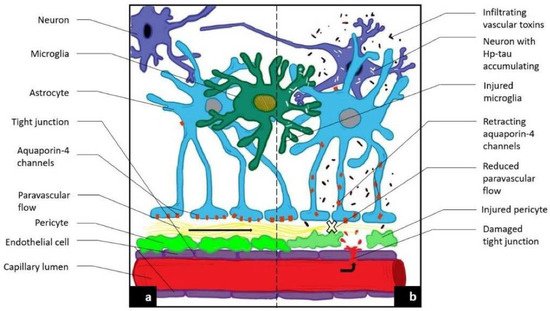
Figure 1. Novel MRI techniques identifying vascular leak and paravascular flow reduction in early Alzheimer disease. (a) Demonstrates normal anatomy and physiology. (b) Demonstrates the pathological progression to late-state AD including the development of a BBB leak through tight junctions, the retraction of aquqporin-4 channels with reduction in paravascular outflow, and accumulation of Aβ and Hp tau.
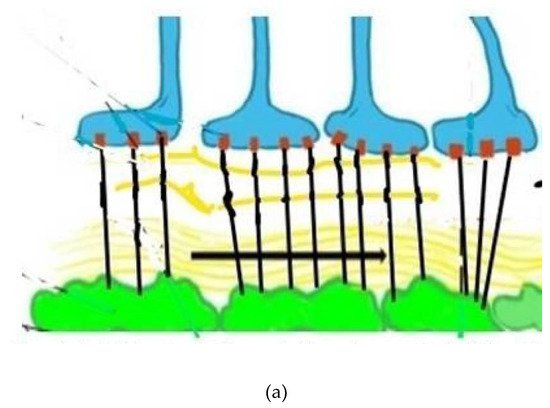
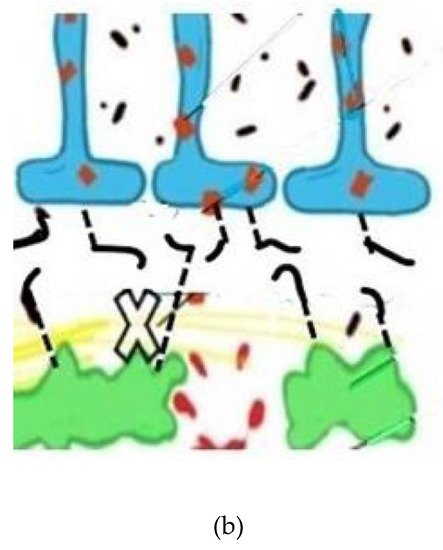
Figure 2. (a) Depicts normal aquaporin attachments in the astrocyte end feet before BBB leak and resulting retraction afterwards. Glymphatic channels and anchoring of the aquaporin 4 channels via pericyte protein attachments. The glymphatic space (white and wavy yellow) which exists between the basement membranes of the astrocytes (blue) and pericytes (green), both surrounding capillary and small venous and arterial vessels (not shown). (b) Depicts damaged pericytes from vascular inflammation and BBB leak with loss of the tethering proteins and retraction of the AQ4 water channels back into the astrocyte soma with resultant loss of glymphatic flow. (Black lines represent tethering proteins and black dots represent leaked restricted proteins). This sequence of disrupted BBB illustrates the initial pathophysiologic damage leading to the leak and sequestration of DAMPS and PAMPS triggering altered proteomic synthesis and degradation and, ultimately, cognitive dysfunction.
The entrance of normally restricted substances into the brain leads to the second stage of disease, the miscleavage of APP probably through intermediate steps and subsequent misfolding of beta amyloid fragments 1–42 followed by crosslinking and extracellular precipitation. Its presence, in time, facilitates production of hyperphosphorylated Tau (hpTau) [19,20,21][19][20][21]. The accumulation of the latter substance is the 3rd and terminal phase of the disease process development [22,23,24][22][23][24]. Because of the deadly nature of hpTau and its propensity to spread transsynaptically as well as by exostosis in a Prion-like fashion, its appearance is the irretrievable end of the road [24]. An apt analogy is considering a pristine lake filled with fish and wildlife fed by a clear stream with waste products draining via an outflow stream. If the water source from the clear stream becomes polluted, over time the lake does as well and the fish begin to die, thus adding to the pollution as well as plugging up the outflow. If one were to simply remove the dead fish but not stop the polluted water inflow, the effect on lake pollution would be minimal, if any, and more wildlife would perish. So, too, removing beta amyloid and hpTau is similar to removing the dead fish and thus, not surprisingly, has little effect on altering disease progression or recovery. It is in the early phase of the disease that repairing the BBB leak must be addressed before the overwhelming effect of toxin induced metabolic disarray and accumulating misfolded proteins develop [25,26][25][26]. To do so requires a reliable method of identifying the altered BBB/glymphatic flow physiology. Testing must identify evidence of fluid leak into the parenchyma and/or reduced outflow via the glymphatic system.
An orderly approach to identifying early sporadic AD includes surveillance of high-risk groups such as those with poorly controlled Diabetes mellitus, hypertension, head injury, morbid obesity, glioblastoma, advanced age and those with chronic inflammatory/infectious processes or identified high risk self-limited infections [27,28,29][27][28][29]. A reliable noninvasive, simple, economic and reliable screening test is desirable to that end.
At this point, it is useful to consider far less common familial AD (accounting for about 5% of all AD), the most common being APOE4 [30,31,32][30][31][32]. If we view these pathways of disease development as being from the inside out as opposed to the outside in (sporadic disease), one must modify the lake analogy to the pollution source being within the lake itself. This may have profound implications as to treatment strategies since stopping the BBB leak would theoretically be less efficacious in the familial group due to the continuous and inherent production of beta amyloid and its negative impact on vascular integrity and the BBB [33,34][33][34]. Having a reliable noninvasive screening test for identifying a BBB leak and glymphatic dysfunction is still necessary even though treatment strategy may differ from sporadic disease. That said, in either case, following the progress of treatment trials’ success requires demonstrating both improvement in glymphatic flow and reduced BBB leak coupled with stable or improved cognitive testing.
2. 3D-ASL Method
Another method is to consider the effect of the BBB breach and its early consequence of reduced glymphatic flow. Arterial spin labeling allows for noninvasive identification of perfusion in and venous flow out of the brain [45,46][35][36]. Utilizing arterial spin labeling and timing the data collection to the post arterial inflow and capillary phases, theoretically, would allow imaging of retained labeled protons primarily within the interstitium and, to a lesser extent, within the venous and glymphatic channels requiring progressively longer inversion intervals from labeling than is used in inflow perfusion. From this, a rate of residual clearance can be determined [46][36]. A reduction in the outflow rate, if present, can be accounted for by both leak in and sequestration of labeled protons within the interstitium and diminished glymphatic outflow [26,46,47][26][36][37]. There are two main obstacles to this approach. The first is knowing the T1 of the environments in which labeled protons will migrate post bolus and the differential flow velocities of arterial, venous and glymphatics [46,47][36][37]. As to the latter concern, choosing time to inversion late in the blood transit cycle reduces contamination from labeled arterial and the majority of venous flow (<2 s/bolus), leaving signal primarily from residual interstitial and glymphatic fluid labeled protons [48,49][38][39]. The T1 at 3T of free fluid is considerably longer (3800 ms) than gray matter (1100–1700 ms), white matter (800–850 ms) or blood (1650 ms), a fact to one’s advantage [49,50][39][40]. The flip side, however, is avoiding the ventricular and subarachnoid spaces in the region of interest (ROI) analysis as this can introduce considerable error. The other major problem with long inversion times is reduced signal to noise (S/N) [51][41].
The transit time of the labeled bolus is about 1.8 s, with contributions to the remaining signal at longer post labeling intervals from mainly labeled fluid in extra-capillary proton environments, which has been measured by other techniques [45,48][35][38]. By intentionally choosing longer post labeling delay times (PLD or TI), signal from retained leaked protons within interstitial fluid and the slower glymphatic flow is maximized. By measuring clearance of fluid outflow, a glymphatic flow rate reduction compared with normal controls is indirect evidence of both a labeled proton leak into the interstitial spaces and their reduced outflow via damaged glymphatics [46][36]. By choosing longer inversion times (in our study, TI’s of 2800–4000 ms at 200 ms intervals past the T1 of blood), the signal contribution of gray and white matter and intravascular elements is minimized and residual labeled brain water signal with the longest T1 is maximized [50,52][40][42]. Because the S/N in these longer inversion times is low, a single determination at any specified time point would result in too much variability for quantitative interpretation [45][35]. By measuring several sequential inversion times, the results can be graphed with the slope of the line indicating the rate of signal loss or clearance from natural signal decay or glymphatic outflow. Linear analysis is possible if the delay times correspond to more linear aspects of the signal decay curve, which can be determined by solving the Block equation for each tissue and fluid component environment within the neuropil [46][36]. Doing so demonstrated that linear analysis in these late delayed signal acquisitions showed high correlation (95%) with the signal decay curve in all relevant tissues and liquid proton environments [46][36]. Thus, residual signal is emitted largely from labeled free fluid protons trapped within the interstitium due to impaired glymphatic outflow [26].
References
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-Brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302.
- Erickson, M.A.; Banks, W.A. Age-Associated Changes in the Immune System and Blood–Brain Barrier Functions. Int. J. Mol. Sci. 2019, 20, 1632.
- Wan, W.; Cao, L.; Liu, L.; Zhang, C.; Kalionis, B.; Tai, X.; Li, Y.; Xia, S. Aβ1–42 oligomer-induced leakage in an in vitro blood–brain barrier model is associated with up-regulation of RAGE and metalloproteinases, and down-regulation. J. Neurochem. 2015, 134, 382–393.
- Fulop, T.; Larbi, A.; Dupuis, G.; Le Page, A.; Frost, E.H.; Cohen, A.A.; Witkowski, J.M.; Franceschi, C. Immunosenescence and Inflamm-Aging As Two Sides of the Same Coin: Friends or Foes? Front. Immunol. 2018, 8, 1960.
- Abbott, N.J. Inflammatory Mediators and Modulation of Blood–Brain Barrier Permeability. Cell. Mol. Neurobiol. 2000, 20, 131–147.
- Thomsen, M.S.; Routhe, L.J.; Moos, T. The vascular basement membrane in the healthy and pathological brain. J. Cereb. Blood Flow Metab. 2017, 37, 3300–3317.
- Tarantini, S.; Tran, C.H.T.; Gordon, G.R.; Ungvari, Z.; Csiszar, A. Impaired neurovascular coupling in aging and Alzheimer’s disease: Contribution of astrocyte dysfunction and endothelial impairment to cognitive decline. Exp. Gerontol. 2017, 94, 52–58. Available online: https://www.sciencedirect.com/science/article/pii/S0531556516303990 (accessed on 22 July 2021).
- Erdő, F.; Denes, L.; de Lange, E. Age-associated physiological and pathological changes at the blood–brain barrier: A review. J. Cereb. Blood Flow Metab. 2017, 37, 4–24.
- Ge, X.; Li, W.; Huang, S.; Yin, Z.; Xu, X.; Chen, F.; Kong, X.; Wang, H.; Zhang, J.; Lei, P. The pathological role of NLRs and AIM2 inflammasome-mediated pyroptosis in damaged blood-brain barrier after traumatic brain injury. Brain Res. 2018, 1697, 10–20.
- Lukiw, W.J. Bacteroides fragilis Lipopolysaccharide and Inflammatory Signaling in Alzheimer’s disease. Front. Microbiol. 2016, 7, 1544.
- Welcome, M.O.; Mastorakis, N.E. Stress-induced blood brain barrier disruption: Molecular mechanisms and signaling pathways. Pharmacol. Res. 2020, 157, 104769.
- Streit, W.J.; Khoshbouei, H.; Bechmann, I. Dystrophic microglia in late-onset Alzheimer’s disease. Glia 2020, 68, 845–854.
- Johnson, E.C.B.; Dammer, E.B.; Duong, D.M.; Ping, L.; Zhou, M.; Yin, L.; Higginbotham, L.A.; Guajardo, A.; White, B.; Troncoso, J.C.; et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med. 2020, 26, 769–780.
- Arellano, J.J.R.; Verkhratsky, A. Neuroglial Roots of Neurodegenerative Diseases? Mol. Neurobiol. 2010, 43, 87–96.
- Kettenmann, H.; Verkhratsky, A. Neuroglia, der lebende Nervenkitt. Fortschr. Neurol. Psychiatr. 2011, 79, 588–597.
- Liu, C.Y.; Yang, Y.; Ju, W.N.; Wang, X.; Zhang, H.L. Emerging roles of astrocytes in neuro-vascular unit and the tripartite synapse with emphasis on reactive gliosis in the context of alzheimer’s disease. Front. Cell. Neurosci. 2018, 12, 193.
- Cohen-Salmon, M.; Slaoui, L.; Mazaré, N.; Gilbert, A.; Oudart, M.; Alvear-Perez, R.; Elorza-Vidal, X.; Chever, O.; Boulay, A. Astrocytes in the regulation of cerebrovascular functions. Glia 2020, 69, 817–841.
- Zenaro, E.; Piacentino, G.; Constantin, G. The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 2016, 107, 41–56.
- Zenaro, E.; Pietronigro, E.; Della Bianca, V.; Piacentino, G.; Marongiu, L.; Budui, S.; Turano, E.; Rossi, B.; Angiari, S.; Dusi, S.; et al. Neutrophils promote Alzheimer’s disease–like pathology and cognitive decline via LFA-1 integrin. Nat. Med. 2015, 21, 880–886.
- Tosun, D.; Landau, S.; Aisen, P.S.; Petersen, R.C.; Mintun, M.; Jagust, W.; Weiner, M.W.; Alzheimer’s Disease Neuroimaging Initiative. Association between tau deposition and antecedent amyloid accumulation rates in normal and early symptomatic individuals. Brain 2017, 140, 1499–1512.
- Jucker, M.; Walker, L. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51.
- Nisbet, R.; Polanco, J.C.; Ittner, L.M.; Götz, J. Tau aggregation and its interplay with amyloid-β. Acta Neuropathol. 2014, 129, 207–220.
- Iqbal, K.; Liu, F.; Gong, C.-X. Tau and neurodegenerative disease: The story so far. Nat. Rev. Neurol. 2015, 12, 15–27.
- Montagne, A.; Zhao, Z.; Zlokovic, B.V. Alzheimer’s disease: A matter of blood–brain barrier dysfunction? J. Exp. Med. 2017, 214, 3151–3169.
- Joseph, C.R. Novel MRI Techniques Identifying Vascular Leak and Paravascular Flow Reduction in Early Alzheimer Disease. Biomedicines 2020, 8, 228.
- Hersi, M.; Irvine, B.; Gupta, P.; Gomes, J.; Birkett, N.; Krewski, D. Risk factors associated with the onset and progression of Alzheimer’s disease: A systematic review of the evidence. NeuroToxicology 2017, 61, 143–187.
- Crous-Bou, M.; Minguillón, C.; Gramunt, N.; Molinuevo, J.L. Alzheimer’s disease prevention: From risk factors to early intervention. Alzheimer’s Res. Ther. 2017, 9, 71.
- Stocker, H.; Möllers, T.; Perna, L.; Brenner, H. The genetic risk of Alzheimer’s disease beyond APOE ε4: Systematic review of Alzheimer’s genetic risk scores. Transl. Psychiatry 2018, 8, 166.
- Montemurro, N.; Perrini, P.; Rapone, B. Clinical risk and overall survival in patients with diabetes mellitus, hyperglycemia and glioblastoma multiforme. A review of the current literature. Int. J. Environ. Res. Public Health 2020, 17, 8501.
- Armstrong, R.A.; Richard, P.; Armstrong, A. Risk factors for Alzheimer’s disease. Folia Neuropathol. 2019, 57, 87–105.
- Tosto, G.; Bird, T.D.; Tsuang, D.; Bennett, D.A.; Boeve, B.F.; Cruchaga, C.; Faber, K.; Foroud, T.M.; Farlow, M.; Goate, A.M.; et al. Polygenic risk scores in familial Alzheimer disease. Neurology 2017, 88, 1180–1186.
- Bell, R.D.; Winkler, E.A.; Singh, I.; Sagare, A.P.; Deane, R.; Wu, Z.; Holtzman, D.M.; Betsholtz, C.; Armulik, A.; Sállström, J.; et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 2012, 485, 512–516.
- Chappell, M.A.; McConnell, F.A.K.; Golay, X.; Günther, M.; Hernandez-Tamames, J.A.; van Osch, M.J.; Asllani, I. Partial volume correction in arterial spin labeling perfusion MRI: A method to disentangle anatomy from physiology or an analysis step too far? NeuroImage 2021, 238, 118236.
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150.
- Joseph, C.R.; Benhatzel, C.M.; Stern, L.J.; Hopper, O.M.; Lockwood, M.D. Pilot study utilizing MRI 3D TGSE PASL (arterial spin labeling) differentiating clearance rates of labeled protons in the CNS of patients with early Alzheimer disease from normal subjects. Magn. Reson. Mater. Phys. Biol. Med. 2020, 33, 559–586.
- Petcharunpaisan, S.; Ramalho, J.; Castillo, M.; Carmichael, O. Arterial spin labeling in neuroimaging. World J. Radiol. 2010, 2, 384–398.
- MacDonald, M.E.; Berman, A.J.L.; Mazerolle, E.L.; Williams, R.J.; Pike, G.B. Modeling hyperoxia-induced bold signal dynamics to estimate cerebral blood flow, volume and mean transit time. NeuroImage 2018, 178, 461–474.
- Okubo, G.; Okada, T.; Yamamoto, A.; Fushimi, Y.; Okada, T.; Murata, K.; Togashi, K. Relationship between aging and T1 relaxation time in deep gray matter: A voxel-based analysis. J. Magn. Reson. Imaging 2017, 46, 724–731.
- Lu, H.; Clingman, C.; Golay, X.; Peter, M.R. Determining the longitudinal relaxation time (T1) of blood at 3.0 Tesla. Magn. Reson. Med. 2004, 52, 679–682. Available online: https://onlinelibrary.wiley.com/doi/abs/10.1002/mrm.20178 (accessed on 22 July 2021).
- Woods, J.G.; Chappell, M.A.; Okell, T.W. A general framework for optimizing arterial spin labeling MRI experiments. Magn. Reson. Med. 2019, 81, 2474–2488.
- Liu, Y.; Zhu, X.; Feinberg, D.; Guenther, M.; Gregori, J.; Weiner, M.W.; Schuff, N. Arterial Spin Labeling MRI Study of Age and Gender Effects on Brain Perfusion Hemodynamics. Magn. Reson. Med. 2012, 68, 912–922.
- Alsop, D.C.; Detre, J.A.; Golay, X.; Günther, M.; Hendrikse, J.; Hernandez-Garcia, L.; Lu, H.; MacIntosh, B.; Parkes, L.M.; Smits, M.; et al. Recommended implementation of arterial spin-labeled perfusion MRI for clinical applications: A consensus of the ISMRM perfusion study group and the European consortium for ASL in dementia. Magn. Reson. Med. 2015, 73, 102–116.
More