NOD-like receptors are functionally diverse intracellular sensors with heterogeneous signaling pathways. With the exception of NLRP10, which lacks an Leucine-rich repeats (LRR) domain, all the containing receptors (NLR) families share a common nucleotide-binding and oligomerization (NACHT) and LRR domain organization.
- NLRP12
- cell death
- inflammasome
- IFN-I
- caspase 1
- NF-κB
1. Introduction
Pathogen recognition, an important initiator of host defense in the early stages of infection and subsequent generation of adaptive immune responses [1][2][1,2], is mediated by a vast repertoire of germline-encoded receptors termed pattern recognition receptors (PRR). These receptors distinguish self from non-self-molecules, i.e., pathogen-associated molecular patterns (PAMPs) [3], such as lipopolysaccharides (LPS), peptidoglycan (PGN), Lipoteichoic acid (LTA), flagellin, microbial nucleic acids, Leishmania Lipophosphoglycan and P. falciparum glycosylphosphatidylinositol (GPI) [4]. The danger/damage-associated molecular patterns (DAMPs), such as extracellular ATP, High mobility group box 1 (HMGB1), parasite hemozoin and uric acid, are recognizable by PRRs and are able to induce inflammatory signaling cascades [5]. Since the earliest report of membrane-bound Toll-like receptors (TLRs) by Jules Hoffman et al. [6] and Charles Janeway Jr [7] in Drosophila melanogaster , as well as the demonstration of canonical toll-like receptors in Cnidarians (Hydra) in recent decades [8], understanding of the immune system and its signal transduction have become more comprehensive. Currently, there are four major classes of PRRs: Toll-like receptors (TLRs), the Nucleotide-binding oligomerization domain (NOD)-Leucine-rich repeats (LRR)-containing receptors (NLR), the retinoic acid-inducible gene 1 (RIG-I)-like receptors (RLR; RIG-I-like helicases—RLH) and the C-type lectin receptors (CLRs). TLRs are considered to be critical in host defense, while non-TLRs are also able to recognize pathogens, to cooperate and regulate the TLRs mediated-signaling cascades that orchestrate the host immune response. Among those non-TLRs, we particularly focus on the NLR family, a series of cytoplasmic sensors with the evolutionarily conserved property, whose roles have rapidly emerged as central regulators of inflammation and immunity associated with relevant human diseases ( Table 1 ). Therefore, the regulation of their activities has become therapeutic targets in most non-infectious and pathogenically-induced inflammatory diseases [9].
| Inflammatory Diseases | Affected Organs | Dysregulated NLRs Family | References | |||
|---|---|---|---|---|---|---|
| Thyroiditis | Thyroid gland | Over-expression and activation of NLRC4, NLRP1, AIM2 and NLRP3 inflammasome | [10][11][12] | [25,26,27] | ||
| Type 1 Diabetes | Pancreas | Over-expression NLRP1, NOD1/2, CIITA and NLRP3 | [11][13][14] | [26,28,29] | ||
| Inflammatory bowel diseases (IBD: Ulcerative colitis and Crohn’s disease) | Gastrointestinal | NOD1, NOD2, NLRP3 and NLRP1 | [11][15][16][17][18] | [26,30,31,32,33] | ||
| Celiac diseases | Small intestine | Enhanced expression of NLRP3 and CIITA, NLRP6 | [11][14][19] | [26,29,34] | ||
| Autoimmune hepatitis | Liver | Hyperactivation of NLRP3 and deficiency of NLRX1 | [2][11][20][21] | [2,26,35,36] | ||
| Arthritis | Joints | Excessive expression of NLRP3, NLRP2, CIITA, NOD2, NLRC5 and NLRP12 (beneficial), and NLRP9 and NLRP11 | [10][11][22][23][24][25] | [25,26,37,38,39,40] | ||
| Systemic Lupus Erythematous (SLE) | Multiple organs such as Kidney, Lung and CNS | Over-expression of NOD2, NLRP3, | SNPs | in CIITA, NLRP1 and NLRX1 | [11][26][27][28][29] | [26,41,42,43,44] |
| Vitiligo | Skin | Increased expression/activation of NLRP1 and NLRP3 | [11][29][30][31] | [26,44,45,46] | ||
| Psoriasis | Epidermal layer (from the limbs to eyelids) | Enhanced expression of NOD2, PYCARD, CARD6, CARD14, NLRP3, NLRP1 and IFI16 | [32][33][34][35] | [47,48,49,50] | ||
| Multiple Sclerosis | CNS: brain, spinal cord and optic nerves. | Over-activation of NOD1, NOD2 and NLRP1. Mutation in CIITA, NLRP3 and regulatory role of NLRP12, NLRC3 and NLRX1 | [36][37][38][39] | [51,52,53,54] |
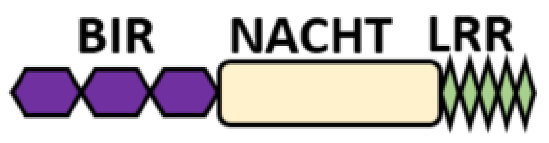
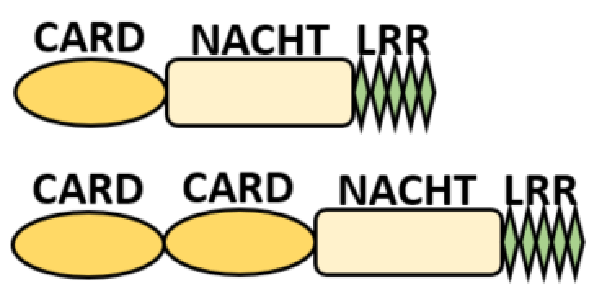
Several homologous NLR genes have been discovered in both the animal and plant kingdoms that demonstrated the plant disease-resistant genes (R gene) encoding nucleotide-binding leucine-rich repeat (NB-LRR) proteins confer the dominant resistance against diverse pathogens, thereby suggesting the conserved biological function of NLR proteins in the host defense [9]. There are approximately 22 and 34 NLR family members in humans and mice, respectively [40][41][42][10,11,12], and lack of specific amino acid sequence in their transmembrane domain identified them as exclusive cytosolic sensors [43][13]. Members of this family share common C-terminal leucine-rich repeat (LRR) domains, a central NOD (nucleotide-binding and oligomerization (NACHT) domain) for ligand recognition and a variable N-terminal effector domain, including caspase recruitment domain (CARD) or pyrin domain (PYD), or the baculoviral inhibitor of apoptosis protein repeat (BIR) domain. These receptors are chiefly expressed by immune cell lineages, such as (macrophages, neutrophils, lymphocytes and dendritic cells), as well as non-hematopoietic cells [44][14]. A myriad of functions have been associated with them beyond the detection of microbial components; they recognize mitochondrial DNA and ATP [45][15], and NLRs regulate TLRs and NOD-derived signaling cascades with the characteristic of adjuvanticity in bacteria and parasitic infections [46][47][16,17]; they are also involved in tissue homeostasis and embryonic development [48][18]. Similarly, NLR proteins have gained much attention in various chronic inflammatory illnesses ( Table 1 ), autophagy [49][19] and carcinogenesis [50][20].
Recently, studies have underscored the impact of NLRs in microbial and parasitic infections. NOD1 and NOD2 play significant roles in the recognition of the bacterial peptidoglycan and provide necessary host defense against Trypanosoma cruzi [51][21] and Plasmodium falciparum [52][22]. Moreover, several species of microbes and parasites have been shown to induce the production of proinflammatory cytokines via NLRP3 and NLRC4 assemblage. The recognition of Listeria monocytogenes by NLRP6 for the recruitment of caspases and GSDMD cleavage has further potentiated NLRs as important molecules in cell death. Similarly, NLRP12 and NLRX1 have been demonstrated to be crucial in the regulation of interferon and cytokine productions, as well as the maintenance of immune homeostasis. The recent discovery of direct activation of noncanonical inflammasome and NLRs by cytosolic lipopolysaccharides (LPS), Lipophosphoglycan (LPG) and Leishmania is critical in programmed cell death (PCD). Moreover, cell death mediated by NLR inflammasome activation is executed to prevent the host cells from pathogen invasion, in which nutrient supply to pathogens is interfered [53][23], and the activated bystander cells could also provide the antimicrobial factors to restrict pathogen expansion and disease progression [54][24]. This review focuses on the significance of NLR proteins in the immune regulation after the recognition of pathogens and the programmed cell death during host defense and immune homeostasis. The potential role of type I interferon in coordinating the inflammasome activation, including the pyroptosis and the latest findings on NLRs as critical checkpoints in host innate immunity, cell death and systemic inflammation, are discussed.
Involvement of the NLRs family in chronic inflammatory diseases.
2. Structure, Function and Classification of NOD-like Receptors
NOD-like receptors are functionally diverse intracellular sensors with heterogeneous signaling pathways. With the exception of NLRP10, which lacks an LRR domain, all the NLRs families share a common NACHT and LRR domain organization. NACHT domain possesses NTPase activity with a binding preference for GTP or ATP [55]. The structural diversity arises from variable N-terminal, which is critical for downstream signaling functions. Therefore, NLRs are phylogenetically divided into the acidic transactivation domain, pyrin domain, caspase recruitment domain (CARD) and baculoviral inhibitory repeat (BIR)-like domains, and NLRX possesses less characterized N-terminal domains usually denoted as X [56] ( Table 2 ).
| NLRs Family | Sub-Family/Domain Architectures | Gene |
|---|---|---|
| Acidic transcription-carrying domain (NLRA) | 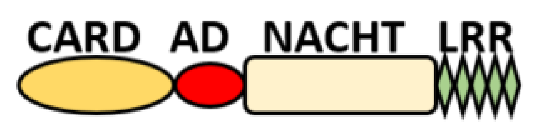 |
CIITA (NLRA) |
| BIR- carrying domain (NLRB) |
 |
NLRB (NAIP) |
| CARD—carrying domain (NLRC) |
 |
NOD1, NLRC4 NOD2 |
| PYD-carrying domain | 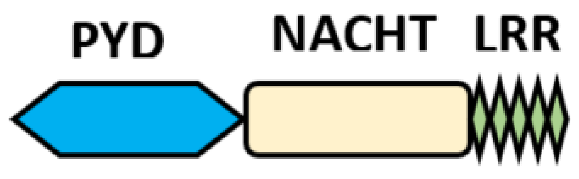 |
NLRP2-NLRP9 NLRP11-NLRP14 |
Additional domain (FIIND) |
NLRP1 | |
No LRR |
NLRP10 | |
| Unidentified domain | NLRs without PYD nor CARD |
NLRC3 NLRC5 NLRX1 |
| NLR-related molecules |  |
Apaf-1 |
Functionally, NOD-like intracellular sensors are classified into four groups;
3. NLRP3 and NLRC4 Inflammasomes
The NLR family pyrin domain-containing protein 3 (NLRP3), a tripartite protein (as described in Table 1 ), and extensively reviewed in the sub-family of inflammasomes [57][58][97,98]. Cellular events, such as ionic flux, the release of ROS, mitochondrial dysfunction and lysosomal damage, have been reported to induce NLRP3 inflammasome activation. Its activation mediates caspase 1-dependent IL-1β and/or IL-18 secretion, and GSDMD dependent pyroptosis. NLRP3 inflammasome comprises NLRP3, adaptor molecule apoptosis-associated speck-like protein containing CARD (ASC, also known as PYCARD) and caspase 1. The oligomerization of NLRP3 at NACHT domains occurs upon stimulation, followed by the recruitment of ASC through hemolytic PYD-PYD. Multiple ASCs merge to further recruit caspase 1 via CARD– CARD interactions to enable self-cleavage and the activation of caspase 1 [57][97]. The activation of NLRP3 can be through canonical and noncanonical pathways [59][99]. Two steps are required in canonical activation: priming/transcription and activation/assemblage, whereas priming is not necessary for noncanonical activation. Priming in the canonical pathway goes through toll-like receptors (TLRs), tumor necrotic receptors (TNFR) [60][100], C-lectin receptors (CLRs) [61][101] and IFN receptors (IFNAR) [62][63][102,103] for the activation of the nuclear factor kappa B (NF-κB) pathway; thus, leading to the transcriptional upregulation of NLRP3 (ready for and post-transcriptional modifications (PTMs), such as ubiquitination and phosphorylation) and pro-IL-1β proteins in the cytosol for ASC recruitment [60][100]. The activation signal for conformation change comes from pathogen and sterile activators (otherwise called PAMPs and DAMPs), examples include: nigericin, extracellular ATP, silica, cholesterol crystals, potassium efflux and reactive oxygen species (ROS). Thereafter, mature caspase 1 will simultaneously induce the cleavage of GSDMD to promote the pyroptotic cell death and the release of mature cytokine proteins IL-1β and IL-18. In contrast, noncanonical activation of NLRP3 bypasses the transcriptional priming and based on direct activation of the noncanonical inflammasome (caspase 4/5/11) in response to the endotoxin LPS from Gram-negative bacteria [64][104], Leishmania Lipophosphoglycan (LPG) [65][105] and oxidized phospholipid1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine(oxPAPC) [66][67][106,107]. Noncanonical inflammasome induces cell death and cleavage of autoinhibited GSDMD into C- and N-terminals; the released N-terminals oligomerize and assemble at the plasma membrane to cause membrane damage and also facilitate potassium efflux and pyroptosis, as well as enhanced NLRP3 inflammasome activation [64][68][104,108]. Therefore, the intrinsic execution of pyroptosis by noncanonical inflammasome requires NLRP3, ASC and caspase 1 for the maturation and secretion of cytokines. Caspase 1 and caspase 4/11 can induce pyroptosis mediated-IL-1β secretion, but caspase 4/11 cannot directly cleave pro-IL-1β and pro-IL-18, but enhance caspase 1 activation for this function instead [69][109]. Recently, there are new reports demonstrating a novel role of caspase 8 and FADD in the regulation of NLRP3 inflammasome activation and maturation of IL-1β [70][71][72][110,111,112]. Kang et al. demonstrate that caspase 8-deficient dendritic cells (DC) exhibit enhanced LPS-induced NLRP3 assembly [73][113], and also similar finding occurred in macrophage during Candida albicans infection [74][114]. However, non-apoptotic caspase 8 seems to play an essential role in TLR signaling-mediated Nlrp3 priming [75][115]. Another study further showed that FADD-caspase 8 is a critical upstream regulator in both canonical and noncanonical NLRP3 inflammasome signaling, as well as in transcriptional priming [76][116]. In macrophages, the deletion of caspase 8 in the presence or absence of RIPK3 inhibited the caspase 1 and caspase 11 activation by NLRP3 stimuli. FADD is positioned upstream of caspase 8, and whose deletion prevents caspase 8 maturation. Mice deficient in FADD and caspase 8 exhibited impaired IL-1β production during C. rodentium infection or while being challenged by LPS [76][116].
NLRP3 inflammasome activation has been overwhelmingly reported to mediate innate immune response in many intracellular pathogens. In gram-positive bacterial infections, NLRP3-dependent caspase 1 regulates the acidification of phagosome buffering by NADPH oxidase NOX2 to modulate innate immune response [77][117]. Hyperactivation of NLRP3 via gain-of-function mutation in the Nlrp3 gene (Nlrp3R258W) promotes the clearance of virus H1N1 IAV infection. This efficacy is ascribed to IL-1β dependent neutrophil recruitment [78][118], but this response was deleterious in the H7N9 viral challenge [79][119]. Pyroptosis mediated by NLRP3 inflammasome plays an essential role in HIV-1–infected patients whose CD4+ T cells are lost [80][120]. This is similar in parasitic infections, such as Trichuriasis [81][121], Neosporosis [82][122], Leishmania [83][84][123,124] , Chagas disease [85][86][125,126], Toxoplasmosis [87][127], Trichomoniasis [88][128] and invasive Entamoeba histolytica [89][129], as well as in fungal infections [90][91][92][130,131,132]. A variety of the corresponding cognate ligands and post-transcriptional modifications exhibited by NLRP3 have been extensively reviewed elsewhere [57][58][97,98]. In parasitic infection, the recognition of Trichuris antigen and exosome by NLRP3 conciliates the secretion of IL-18 that promotes parasite persistence. Nlrp3 −/− mice with reduced proinflammatory type 1 cytokine responses and augmented protective type 2 immunity results in worm expulsion [81][121]. Extracellular Entamoeba histolytica also induces NLRP3-dependent caspase 1 via its contact with α 5β 1 integrin at the Macrophage- Amebae intercellular junction. The α 5β 1 integrin induced ATP release into the extracellular space through the opening of pannexin-1 channels that signaled through P2X 7 receptors to deliver a critical co-stimulatory signal that activates the NLRP3 inflammasome for the induction of IL-1β [89][129]. Likewise, ROS production was induced after Neospora caninum to mediate NLRP3 activation and the production of proinflammatory cytokine in macrophages [82][122].
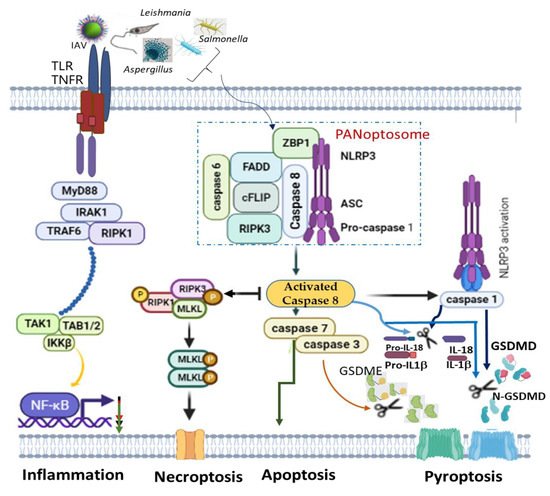
A recent finding on PANoptosis (concomitant activation of three autonomous cell deaths: pyroptosis, apoptosis and necroptosis) also identified NLRP3 as one of the indispensable components of PANoptosome ( Figure 1 ). PANoptosis is a programmed cell death (PCD) common in macrophages infected with intracellular pathogens, such as influenza A virus, vesicular stomatitis virus, Listeria monocytogenes , Salmonella enteric serovar Typhimurium , Candida albicans and Aspergillus fumigatus , to circumvent pathogen-mediated inhibition [93][94][133,134]. The recognition of pathogens or their products by ZBP1 mediates the assemblage of a cytoplasmic multimeric protein complex, known as PANoptosome, and induces PCD called PANoptosis. This complex comprises NLRP3, ASC, CASP8, RIPK3, CASP6 and Z-DNA binding protein 1 (ZBP1) [94][95][96][134,135,136]. ZBP1, which is a DNA-dependent activator of IFN-regulatory factors (DAI) [97][137], interacts with NLRP3 inflammasome, and together with CASP8, to form a PANoptosome [98][99][138,139]. Simultaneous deletion of NLRP3 inflammasome components and caspase 8 largely rescued multiple PCDs than an individual deletion, which provided reduced or no redemption of PCD in pathogen-infected macrophages [100][140]. This is simply because caspase 8 can exhibit non-apoptotic function to directly cleave pro-IL-1β, pro-IL-18 and GSDMD as well as promotes GSDME-mediated pyroptosis via downstream caspase 3 and caspase 7 activations [96][136]. However, even though caspase 8 is an essential modulator of PANoptosis, its shifting towards necroptosis and pyroptosis is possible alongside RIPK3 and NLRP3 activation, respectively [101][102][141,142] ( Figure 1 ).
NLRC4 , NLR family CARD domain containing 4 formerly called IPAF (ICE protease-activating factor) for its ability to activate caspase 1, is another canonical inflammasome that is tightly regulated by transcriptional and posttranscriptional mechanisms. Its expression is upregulated by TNF and the stress-mediated p53 activation [103][104][105][143,144,145]. NLRC4 contains a CARD domain, and therefore, can directly recruit pro-caspase 1 via CARD– CARD interaction to activate caspase 1, associates or co-localizes with ASC [103][106][143,146] for proteolytic cleavage of proinflammatory cytokines (pro–IL-1β and pro–IL-18) and GSDMD [107][108][147,148]. The pathogen recognition of NLRC4 is usually via a sensor NAIP (NLR family, apoptosis inhibitory proteins) [109][149]. Mouse NAIP5/NAIP6, NAIP2 and NAIP1 specifically recognize bacterial flagellins, TTSS rod and T3SS needle proteins, respectively, to induce the activation of NLRC4 inflammasome [109][110][111][149,150,151]. Human NAIP merely recognizes T3SS rod protein PrgJ [112][152], and its splice variant senses flagellin to promote NLRC4 inflammasome activation in humans [113][153]. The NLRC4-dependent caspase 1 activation and cell death are cell-specific. NLRC4 activation is critical in macrophages infected by intracellular invasive bacteria Salmonella typhimurium and Burkholderia thailandensis , which mediates pyroptotic cell death and the release of proinflammatory cytokines: IL-1β and IL-18 [114][115][116][154,155,156]. However, unlike macrophages, NLRC4 activation in neutrophils was found to be critical for caspase 1-dependent IL-1β production but not pyroptotic cell death [64][114][104,154]. Pyroptotic cell death in neutrophils is majorly coordinated by noncanonical inflammasomes (caspase 4/5/11). A study also revealed that NLRC4 mediates IL-18 secretion necessary to drive IFN-γ, which subsequently primes both macrophages and neutrophils for caspase 11 activation during Burkholderia thailandensis infection in mice [115][155]. In addition, leucine-rich repeat kinase 2 LRRK2 is an intrinsic regulator of NLRC4 and LRRK2 formed a complex with NLRC4 for its optimal phosphorylation at Ser533, which promotes inflammasome activation during S. typhimurium infection in macrophages [117][157]. Interestingly, just as a report showed, acetylation-induced NLRP3 activation can be reversed by SIRT2 [118][158]; also SIRT3 was found to promote NLRC4 inflammasome activation by deacetylation [108][148]. The involvement of NLRC4 in pathogen recognition was largely thought to be restricted to bacterial infections but recent studies revealed that dendritic cell NLRC4 regulates T cell response during influenza A virus infection. Similarly, NLRC4 was demonstrated to promote the susceptibility of Paracoccidioides brasiliensis (pathogenic fungus infection) by regulating NLRP3 activities to dampen the late IL-18 production and CD8+ IFN-γ+ T cell responses [119][159].
4. NOD-Like Receptors in the Regulation of Pyroptosis Cell Death
Pyroptosis is a lytic programmed cell death (PCD) that involves cell swelling and the rupturing of the plasma membrane, and it remains a major pathway for the release of proinflammatory proteins in macrophages [120][207], dendritic cells and partially in neutrophils [121][122][208,209]. This cell death was first observed by Friedlander et al. in 1986 in primary mouse macrophages with anthrax lethal toxin (LT) treatment leading to the rapid release of cell contents [123][210] and later established in Shigella flexneri -infected macrophages by Zychlinsky and his co-workers in 1922 [124][211].
The establishment occurred after the discovery of ICE (interleukin-1β-converting enzyme) otherwise called caspase 1 [125][212] as a critical weapon for the maturation of IL-1β from the precursor [126][127][213,214]. However, the phenomenon was first thought to be apoptosis until 2001 when D’Souza et al. coined the term pyroptosis as proinflammatory programmed cell death to distinguish it from non-inflammatory cell death called apoptosis [128][129][215,216].
The discovery of inflammasome in 2002 [130][217] made it clearer that efficient execution of pyroptosis requires the activations of the intracellular inflammasome signaling complex to activate inflammatory caspase 1 necessary for the process of IL-1β and IL-18 from pro-IL-1β and pro-IL-18, respectively. Until 2015, when gasdermin D was discovered as a substrate target of caspase 1/4/5/11, the pyroptosis effector molecule was unknown [131][132][218,219]. The oligomerization of the cleaved active form of gasdermin protein (N-terminal of GSDMD) [130][131][217,218] results in the lysis of cells and facilitates the release of proinflammatory cytokines (such as IL-1β, IL-18 and TNF) and alarmins, such as high mobility group box 1 (HMGB1) [120][121][207,208]. It is noteworthy to mention that the first report of the gasdermin gene (now GSDMA) in the gastrointestinal tract (tightly restricted to the esophagus and stomach) and skin of a mouse was 2000 [133][220], and since then, many members of the gasdermin family have been reported; GSDMB, GSDMC, GSDMD, GSDME (also known as DFNA5) and PJVK (also known as DFNB59), with different activating enzymes both in human and mouse [134][135][136][221,222,223]. This expansive advancement in the understanding of the gasdermin family and its activating protease enzymes, such as inflammatory caspase 4/5/11 [72][134][112,221], non-inflammatory caspase 3/7/8 [135][136][222,223], Cathepsin G [137][224] and neutrophil elastase [138][225], as well as inflammasome activation [139][140][226,227], has further broadened the concept of cell death as critical therapeutic targets [141][142][228,229] in host immunity [143][144][230,231], microbial-induced hyperinflammation [100][145][140,232], cytokine storm syndrome [141][228] and autoimmune diseases [122][146][209,233], as well as in severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [141][147][228,234]. Today, program cell deaths (PCD), particularly pyroptosis along with others, such as Necroptosis [148][235], Ferroptosis [149][236], NETosis [64][150][104,237], Parthanatos [151][238] and PANoptosis [93][95][133,135], have received a lot of attention from all facets of research disciplines. Fortunately, in spite of scrupulous attention to unravel the underlying molecular mechanism, the phenomenon of immunological cell death is still progressively complicated [152][239]. Considering the impact of cell death in microbial (viral, bacterial and fungal) and parasitic infections, PCD appears to be a double-edged sword for host and pathogen survival [153][240]. Therefore, tight regulation of the phenomenon is crucial for immune homeostasis. In this part of the review, we focus on the interplay of the afore-listed group of NLRs (trans-activator, inflammasome and regulatory NLRs) and other critical molecules, such as type 1 interferon (IFN-I) and inflammatory caspases ( Caspase 1/4/5/11), on the initiation, execution and regulation of pyroptosis ( Figure 2 ).
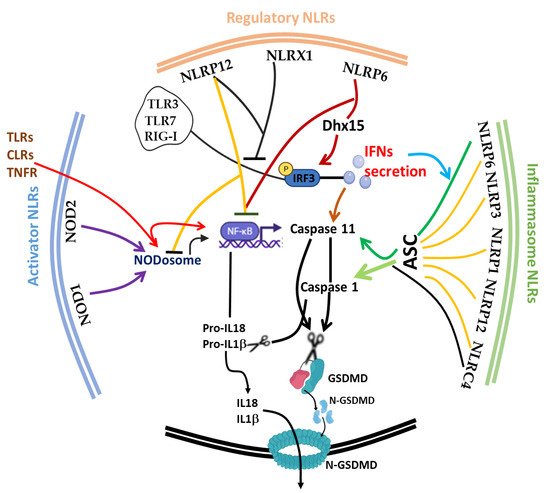
During microbial and parasitic infections, regulated cell death (RCD) [63][103] mediated by appropriate secretion of IFN-I [154][242] and activation of the inflammasome is vital for the host to cope with either foreign pathogens or tissue damage. Uncontrolled activities of these players can cause aberrant tissue damage, autoinflammatory disorders, cardiometabolic diseases, cancer and neurodegenerative diseases. IFN-I was previously recognized as a crucial molecule that is involved in the protection against viral infections [155][156][243,244]; the current paradigm shift has shown its impact on a range of microbial infections, such as parasites, fungi and bacteria. Similar to cell death, IFN-I is also a double-edged sword that exhibits context-dependent functions in relation to the intrinsic and extrinsic factors in cells [154][157][242,245]. Protective host defense of IFN-I was demonstrated in Acinetobacter baumannii [158][246], Escherichia coli [159][247], Helicobacter pylori [160][91], Legionella pneumophila [161][248] Mycobacterium abscesssus [162][249], Plasmodium berghei [163][250] and Aspergillus species, A. fumigatus , A. nidulans and A. tanneri [164][251]. Other reports also shown that it promotes detrimental infection outcome in bacteria, such as Chlamydia muridarum [165][252], Mycobacterium bovis [166][253], Escherichia coli [167][254] , Francisella tularensis [168][255], Haemophilus influenza [169][256] and Salmonella typhimurium -viral [170][257], likewise in fungal and parasitic infections, such as Candidiasis [171][258], as well as in Chagas parasitic infection caused by Trypanosome cruzi [172][259]. In Listeria monocytogenes [173][174][260,261], Pseudomonas aeruginosa [175][176][262,263], Salmonella enterica serovar Typhimurium [177][178][264,265] and Yersinia pestis [179][266] , IFN-I confers dual functions depending on the IFN-stimulated genes (ISGs) involved and the route of infection. For instance, contrary to what is obtainable during foodborne transmission of listeriosis, IFN-I promotes bacterial susceptibility during intravenous route infection by creating a growth-tolerable intracellular microenvironment [174][261]. Mechanistically, Frantz et al. and Pagliuso et al. recently and respectively described secRNome and RNA-binding protein (Zea) secreted by L. monocytogenes were sensed by RIG-I to mediate IFN-I secretion and signaling pathways [173][180][260,267], and this is explored by bacteria to its advantage.