Cystic fibrosis (CF) is the most common of rare hereditary diseases in Caucasians, and it is estimated to affect 75,000 patients globally. CF is a complex disease due to the multiplicity of mutations found in the CF transmembrane conductance regulator (CFTR) gene causing the CFTR protein to become dysfunctional. Although CFTR is the main chloride channel in the lungs, others could, e.g., anoctamin-1 (ANO1 or TMEM16A), compensate for the deficiency of CFTR.
- cystic fibrosis
- anoctamin-1
- calcium-activated chloride channel
- CFTR-independent therapy
1. Introduction
2. Anoctamin-1
3. ANO1 in Cystic Fibrosis
3.1. Overview
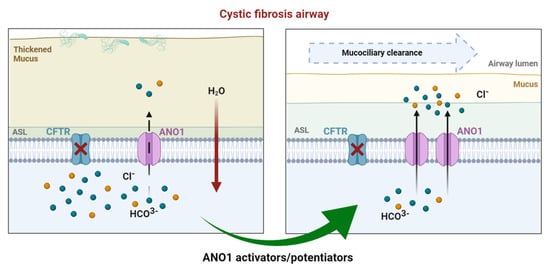
3.2. Drug Approaches Targeting ANO1 in Cystic Fibrosis
Inhibitors | Specificity | Assay | References | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
ANI9 | Not specific | In vitro | |||||||||
CCinh-A01 | Not specific | In vitro, in vivo |
[26] |
[86] |
|||||||
DIDS | Not specific | In vitro |
[27] |
[87] |
|||||||
Diphenylamine-2-carboxylate (DPC), 5-nitro-2-(3-phenylpropylamino) benzoic acid | Not specific | In vitro |
[28] |
[88] |
|||||||
Flufenamic acid | Not specific | In vitro | |||||||||
Niclosamide | Not specific | In vitro, in vivo |
[31] |
[91] |
|||||||
Niflumic acid | Not specific | In vitro, in vivo |
[32] |
[92] |
|||||||
Plumbagin | Not specific | In vitro |
[33] |
[93] |
|||||||
Quercetin | Not specific | In vitro, in vivo, clinical trial (phase II) | |||||||||
Tannic acid | Not specific | In vitro |
[37] |
[97] |
|||||||
T16ainh-A01 | Specific | In vitro |
[26] |
[86] |
|||||||
Activators | |||||||||||
Denufosol (INS37217) | Not specific | In vitro, in vivo, clinical trial (phase III failed) | |||||||||
Duramycine (MOLI1901) | Not specific | In vitro, in vivo, clinical trial (phase II failed) |
[45] |
[100] |
|||||||
Eact | Not specific | In vitro |
[26] |
[86] |
|||||||
ETD002 (or ETX001) | Specific | In vitro, in vivo, clinical trial (phase I) |
[46] |
[101] |
|||||||
Interleukin 4 | Not specific | In vitro |
[14] |
||||||||
Resveratrol | Not specific | In vitro, in vivo, clinical trial | |||||||||
TSB ANO1 | Specific | In vitro, in vivo, preclinical |
[50] |
[83] |