Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Jakub Litak and Version 2 by Catherine Yang.
Glioblastoma (GBM) is the most popular primary central nervous system cancer and has an extremely expansive course. Aggressive tumor growth correlates with short median overall survival (OS) oscillating between 14 and 17 months. The survival rate of patients in a three-year follow up oscillates around 10%. The interaction of the proteins programmed death-1 (PD-1) and programmed cell death ligand (PD-L1) creates an immunoregulatory axis promoting invasion of glioblastoma multiforme cells in the brain tissue. The PD-1 pathway maintains immunological homeostasis and protects against autoimmunity. PD-L1 expression on glioblastoma surface promotes PD-1 receptor activation in microglia, resulting in the negative regulation of T cell responses.
- PD1
- PD1 ligand
- glioblastoma multiforme
1. Introduction
Glioblastoma (GBM) is the most common primary cancer in the central nervous system and has an extremely expansive course [1]. The standard approach to glioma treatment consists in the most extensive as possible surgical resection and in adjuvant radiation strengthened by temozolomide (TMZ) administration [2]. The median overall survival (OS) oscillates between 14 and 17 months [3][4][3,4]. The survival rate of patients in a three-year follow up oscillates around 10% [5]. GBM resistance to typical therapies requires verification. Glioblastoma cells interact with the surrounding environment, creating forceful interactions among heterogenous cell groups, various chemokines with cytogenetic effects, and extracellular proteins stimulating tumorigenesis, uncontrolled multifocal expansion, and immunological evasion [6].
The proteins programmed death-1 (PD-1) and programmed cell death ligand (PD-L1) interplay, creating an immunoregulatory axis promoting invasion of glioblastoma multiforme cells in the brain tissue [7]. Physiologically, the main function of PD-1 is to restrain T-cell anti-tumor activity and amplify Tregs activation, which limit T-cell reaction and protect against hyper immunity. The PD-L1/PD-1 axis maintains immunological homeostasis and protects against autoimmunity [8].
2. PD-L1 and PD-1 Structure
PD-L1 is encoded by the gene PDCDL1, localized on the 9th chromosome in p24.1 position. PD-L1 is also described as CD274 and B7-H1. This ligand was discovered and described in 1999 by Dong et al. as a member of the B7 protein family [9][17]. Seven exons encode the full-length protein PD-L1, consisting of 290 amino acids (40 skDa). PD-L1 as a type-I transmembrane complex of proteins includes single IgV and IgC domains on the external part, a transmembrane domain with hydrophobic properties, and a 30-amino acid cytoplasmatic tail as a signal transducer [10][18]. PD-L1 activity depends on binding to the PD-1 receptor encoded by the PDCD1 gene. It is information transcribed from the second chromosome and consists of 288 amino acids (50–55 kDa). It contains an IgV domain in the extracellular domain and transmembrane region [11][19]. The intracellular region forms a tail composed of a tyrosine-based switch motif (ITSM)–inhibitory motif. This receptor was described by Ishida et al., who used subtractive hybridization to identify genes regulating programmed cell death [12][20]. PD-L1 is expressed and excreted by neoplastic cells, APCs, lymphocytes B, and parenchymal cells. It induces T-cell apoptosis or anergy and modulates inflammation in situ [13][14][21,22]. The binding of PD-1 to the corresponding PD-1 receptor activates the protein tyrosine phosphatase SHP-2, which dephosphorylates Zap 70 (Figure 1). This process T cells proliferation and downregulates lymphocyte cytotoxic activity [15][23].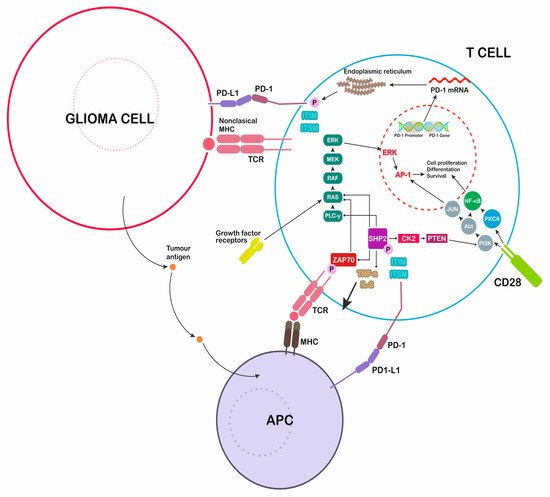
Figure 1. Immunological modulation induced by glioblastoma multiforme (GBM) cells. Secretion of programmed cell death ligand 1 (PD-L1) inhibits the immune attack, blocking T cells responses. PD-1: programmed death-1, major histocompatibility complex: MHC, T-cell receptor: TCR, antigen-presenting cell: APC.
3. PD-1 Ligand Expression—Role of TLR Activation
Toll-like receptors (TLRs) and agonists of receptors induce the immune response, activating many pathways, and cooperate with various antigens. TLRs are a conserved family of 10 receptors (TLR1–10) taking part in pattern recognition [16][27]. Agonists of this group of receptors used to be called pathogen-associated molecular patterns (PAMPs). Their binding to specific TLRs initiates an immune response [17][18][28,29]. Studies revealed that TLRs are endogenously expressed in glioma cells. The fact that TLR2, TLR4, and TLR9 activated by agonists promote tumor expansion and proliferation complicates the role of TLRs in the antitumor response [19][30].
TLR agonists as microbial antigens, activating TLR receptors to initiate precise immunological activities. Agonists of TLR that have been studied include lipopeptides (TLR2, TLR6, and TLR1 agonists), lipopolysaccharides (TLR4 agonist), LPS, flagellin (agonists of TLR5), single-stranded DNA (TLR8 agonist and TLR7), double-stranded (ds)DNA (agonist of TLR3), and the DNA CpG motif (agonist of TLR9). Later studies showed that autocrine molecules released from dead and stressed cells such as heat shock proteins (HSP, for TLR4 and TLR2) and high-mobility group box 1 proteins (HMGB1, for TLR4 and TLR2) are also significant agonists. Many of them appear in glioma environment, causing tumor induced-activity of TLRs [20][21][31,32].
In GBM cells, constitutive elevated expression of αB-crystallin, HSP27, HSP73, HSP72, and HSP90 was reported in vivo and in vitro as a result of endogenous induction. HSP–peptide complexes (HSPPCs) are able to interfere with various superficial receptors such as CD36, CD91, CD40, CD14, TLR2, TLR4 [22][23][24][33,34,35].
TLR activation in glioma cell results in signaling through two main pathways, one of which is myeloid differentiation factor 88-independent (MyD88-independent), and the other is myeloid differentiation factor 88-dependent (MyD88-dependent) (Figure 2). The MyD88-dependent signaling cascade (MyD88/TRAF6/MEK/ERK) promotes early activation of cytokine transcription through NF-κB, supporting inflammatory processes and cytosolic enzyme and chemokine activity, and starts PD-L1 gene transcription. The MyD88-independent pathway leads to the late activation of NF-κB and interferon regulatory factors (IRF), which control the expression of type I IFNs and the activation of many gene promoters. Excreted Type I IFNs influence PD-L1 overexpression through IFNAR signaling activation, proving an indirect effect of the independent pathway [25][26][27][36,37,38].
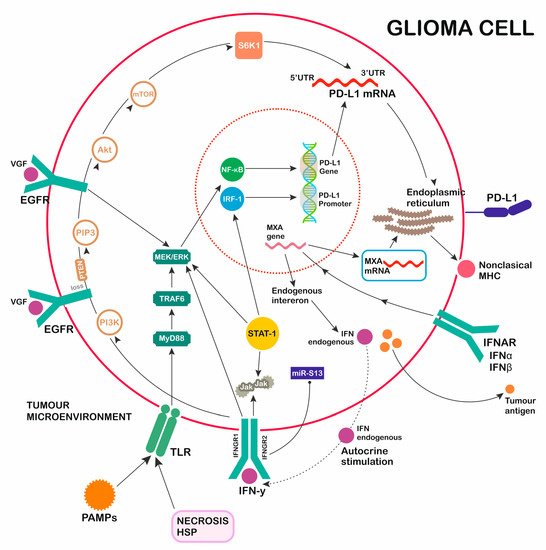
Figure 2. GBM induction of PD-L1 secretion. Multiple activation pathways (TLR, EGFR, IFNAR, IFNGR) promoting PD-L1 expression. 1. Toll-like receptors (TLR) pathway: pathogen-associated molecular patterns (PAMPs), NECROSIS, heat shock proteins (HSP) as activators of TLR myeloid differentiation factor 88 (MyD88)-dependent pathway signaling through TRAF6/MEK/ERK/NF-κB. 2. Epidermal growth factor (EGFR) pathway: TGFα/EGF/VGF/MUTATION OF RECEPTOR as activators of EGFR pathway signaling through MEK/ERK (STAT-1)/NF-κB. 3. IFNAR pathway: interferon (IFN)alfa, IFNbeta as activators of IFNAR pathway signaling through MXA gene transcription, forming nonclassical MHC, promoting PD-1L transcription, and the induction of endogenous interferons. 4. IFNGR pathway: IFNgamma as an activator of IIFNGR pathway signaling through JAK/STAT-1/MEK/ERK/IRF-1 and PI3K/PIP3/Akt/mTOR/S6K1, with a regulatory function over transcribed PD-1L mRNA.