Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Amedeo Amedei and Version 2 by Peter Tang.
Obesity is a multifactorial disorder in which various elements (genetic, host, and environment), play a definite role, even if none of them satisfactorily explains its etiology. A number of neurological comorbidities, such as anxiety and depression, charges the global obesity burden, and evidence suggests the hypothesis that the brain could be the seat of the initial malfunction leading to obesity. The gut microbiome plays an important role in energy homeostasis regulating energy harvesting, fat deposition, as well as feeding behavior and appetite. Dietary patterns, like the Western diet, are known to be a major cause of the obesity epidemic, probably promoting a dysbiotic drift in the gut microbiota.
- obesity
- microbiota
- gut–brain axis
- neurological disorders
- nervous system
- inflammation
1. Introduction
Obesity is an abnormal or excessive fat mass accumulation that affects the health status. The worldwide epidemic of obesity has become an important public health issue, with serious psychological and social consequences since, worldwide, over 650 million adults and 340 million children and adolescents are obese [1]. Obese phenotypes can be associated with some genetic predispositions [2][3][2,3] and with sedentary lifestyle. However, these factors alone fail to accurately describe and explain the complexity of the phenomenon. Obesity is a multifactorial disorder which is a result of the interaction of host and environmental factors and its prevalence in high income and upper middle-income countries is more than double that of low and lower middle income countries [4]. Because of that, it constitutes a social problem, especially in those upper middle-income countries, not just affecting the welfare state, but also creating issues in terms of social relations and acceptance, and personal development. Moreover, obesity is considered, sometimes erroneously, as the consequence of an unbalanced and/or mistaken feeding conduct. On the contrary, recent long-term studies reveal a more complex scenario, including neuropsychological and neurobiological factors [5], that in turn involve not only a different categorization of the pathology itself but also suggest that obesity cannot be adequately treated through simple nutritional plans, associated with training and exercise [6]. By considering the complex nature of the pathology (from genetic factors to behavioral and social ones) and given the poor effectiveness of many pharmacological and nutritional approaches, some researchers suggest that “behavioral dimension” should not be neglected, in order to develop new approaches, both preventive and therapeutic, that include obesity within neuropsychological syndromes [5].
2. Why a Neuropsychological View of Obesity?
Obesity occurs when energy intake exceeds energy expenditure over time, and, traditionally, it is considered as the consequence of a sedentary lifestyle and the usual excessive food consumption. Excessive adiposity is a major risk factor for cardiovascular disease, cancer, type 2 diabetes, and mood-related disorders, with obese individuals often suffering social stigmatization [7][8][16,17]. Given its multifactoriality, the obesity is a complex disease in which both genetic and environmental factors play a role in its development. Anyway, none of them satisfactorily explains the etiology yet and many details of this pathological condition remain murky. Since differences in the brain could be both a consequence of, and/or an explanatory factor for obesity, recent attention has shifted towards its neurobiological features, in particular in the pathogenic processes and in the clinical-related neurological conditions.3. The Microbiota–Gut–Brain Axis
The intestine and the brain are intimately connected by the gut–brain axis, a complex bidirectional system in which the central and enteric nervous system communicate involving endocrine, immune and neuronal pathways. Communication and functions of this axis are regulated by the GM at the point that the concept of microbiota–gut–brain (MGB) axis has been introduced to underline the pivotal role of GM in the development of metabolic and neurological diseases.
The microbiota represents the community of microbes (bacteria, archaea, viruses, and fungi) that reside in a particular habitat (e.g., the gut microbiota) and establish with the host a mutually beneficial relationship (while the “microbiome” represents the collective genomes of microorganisms) [9][103]. In particular, the microbiota offers benefits to the host maintaining the gut integrity [10][104], harvesting energy [11][105], providing protection against pathogens [12][106] and regulating the immune system [13][107]. The human gastrointestinal tract holds more than 1000 bacterial species, mainly located within distal ileum and colon, which belong prevalently from Bacteroidetes and Firmicutes phyla. The GM com-position is highly dynamic and susceptible to rapid changes in response to external factors as diet, stress, smoking, infections, or perturbation of the healthy state [14][108]. In turn, changes in GM com-position and function, named dysbiosis, can be responsible for the development of various diseases (e.g., colorectal cancer) [15][16][109,110], and, can contribute to the disruption of the molecular dialogue between gut and brain [17][111].
The MGB axis is composed of the CNS, the autonomous nervous system (ANS), the neurons of the enteric nervous system (ENS), the HPA-axis and the GM. In particular, signals from the brain influence the motor, sensory, and secretory modalities of the gastro-intestinal tract, regulate the inflammatory process and influence the GM structure [18][112]. In turn, visceral messages from the gastro-intestinal trait can influence brain function [18][19][112,113]. For instance, under stress conditions, the cortisol released following HPA axis activation alters the gut permeability and barrier function, thus affecting the GM composition [18][112].
Conversely, the gut microbiome influences the brain functions modulating the levels of various brain transmitters (i.e., serotonin) [20][114] and circulating cytokines, that can exceed the BBB [21][22][115,116].
Growing evidence, involving studies in germ-free (GF) animal models, which intestinal flora is missing from birth, and humans exposed to probiotic agents or antibiotics, suggests that several pathologic conditions may be affected by a MGB axis dysregulation, such as autism spectrum disorders [23][24][25][117,118,119], anxiety/depression [26][120], and obesity [27][28][29][121,122,123].
3.1. Microbiome and Energy Harvest
The intestinal microbiome plays a key role in digestion and absorption of nutrients, regulating energy homeostasis through different mechanisms as energy extraction from food and the modulation of fat storage by the short chain fatty acids (SCFAs) and monosaccharides absorption.
The first information about the role of bacterial flora in the obesity physiopathology was obtained in GF mouse models [30][31][32][124,125,126]. These mice were significantly leaner than controls, despite introducing more calories from food [33][127]. Moreover, GF mice showed modified plasma fatty metabolic markers and lower amount of leptin and ghrelin, suggesting an energy imbalance [34][128]. When GF mice were transplanted with gut bacterial flora obtained from conventionally raised mice, they showed an increase in insulin resistance and in body fat without an observed increment in nutrient intake [29][123]. This evidence has placed the gut microbiome at the center of a completely new research field, concerning the pathophysiology of obesity. Biochemical and metagenomics data analysis suggested that the “obese microbiota” [35][14] was able to harvest more energy from the diet and this ability was also transferable. Thus, the colonization of GF mice with an obese microbiota (human or murine) induced an increase in total body fat higher than that one obtained through colonization with a lean microbiota [36][129]. In addition, when eutrophic GF mice received fecal microbiota from obese women, metabolic complications associated to obesity have been observed [37][130]. This evidence indicates how rapid, transmissible and flexible can be the relation between food and commensal microorganisms in obesity and metabolic syndrome.
Animal obesity models and obese humans present a similar microbial phylum taxonomic rank dysbiosis. In particular, humans and mice share two main phylum: Firmicutes and Bacteroidetes; an alteration of Firmicutes/Bacteroidetes ratio was observed in several obesity studies in both mice [38][131] and humans [35][14]. Indeed, the Firmicutes could break down indigestible carbohydrates and converting them into absorbable energy products [39][40][41][132,133,134]. However, in a meta-analysis study the observed alteration in the ratio Firmicutes/Bacteroidetes seemed to be unrelated to weight differences [42][135]. Also, the reduction in microbial diversity and alteration of particular microbial families or species have been observed in obesity conditions [20][114], such as the increase of Proteobacteria [43][136].
The complex interplay between host genetics, gut microbiome and environmental factors is crucial for the obesity pathophysiology (for instance, monozygotic twins showed a more similar GM profile than did dizygotic twins) [44][137]. Dietary pattern could also affect the bacterial structure, for example, westernized diets increase the abundance of Clostridia (Firmicutes phylum) populations that could extract more energy from the diet, consenting higher energy utilization [45][138]. The unused extra energy is then accumulated as fat deposits.
While different processes, by which an ‘‘obese microbiota’’ can affect body weight balance [33][46][47][127,139,140] have been indicated, the increased energy harvest via colonic fermentation and SCFAs’ production is the most direct [35][14]. Bacterial enzymes (specific glycoside hydrolases) metabolize otherwise not digested by humans food components, like vegetable fibers (such as resistant starch, cellulose, and inulin) that cannot be metabolized by human enzymes. The final product of this process are energy-rich substrates, such as SCFAs [48][141]. SCFAs can provide ≤10% of total daily caloric intake [49][142]. Obese individuals show significantly increased levels of SCFAs such as acetate, propionate, and butyrate [40][50][133,143]. Most of the bacterial SCFAs (in particular butyrate) are derived from the fermentation process of Clostridia cluster [51][144]. SCFAs not only operate as energy substrates for host tissues and bacteria but also act as signaling molecules in the host metabolism, showing a relevant role in mediation of gut motility, regulation of fat storage and appetite [11][105]. Indeed, dysbiosis induced by the type of diet has been correlated with an acetate increase that promotes hyperinsulinemia [52][145]. Furthermore, SCFAs can influence other obesity-associated conditions such as insulin resistance and hyperglycaemia [53][146].
Among the SCFAs, the propionate can be utilized locally through conversion into glucose by intestinal gluconeogenesis or diffuse into the portal vein to be utilized as a substrate for hepatic gluconeogenesis, preventing high SCFAs concentrations in blood [54][147]. In addition, propionate decreases human lipogenesis and serum cholesterol (in hepatic and non hepatic tissues) [55][148], and also reduce the fasting blood glucose and hepatic cholesterol in obese rats [56][149].
In addition, several studies shows acetate benefits on metabolism. Acetate could bind to the receptor GPR43 in several target organs. In adipose tissue, the GPR43 activation inhibits insulin signaling and suppresses fat accumulation, while systemically, it improves insulin sensitivity [57][150]. Mice deficient in the acetate receptor GPR43 become obese when fed a normal diet, whereas mice who overexpress GPR43 remain lean even when fed an obesogenic diet [57][150]. In the liver, acetate reduces lipid accumulation and improves liver function and mitochondrial efficiency. In adipose tissue, acetate inhibits fat breakdown but induces the browning of white adipose tissue and metabolic improvements, leading to a reduction in body fat [58][151]. Finally, prebiotics such as inulin, increased acetate production that crosses the blood–brain barrier of rats and results in reduced grehlin production and so, inducing a decrease of body weight, food intake, and fat mass [59][152]. Moreover prebiotic fructooligosaccharides increase acetate production, reducing body weight and fat mass because it favors a lower food intake in mice [60][153].
Microbial metabolites can also regulate the composition of bile acid species. A reduced bile acid amount in the intestine has been associated with inflammation and microbiota overgrowth [61][154]. Some intestinal bacteria are able to extract energy from the metabolization of bile acids, inducing the activation of bile acid receptors farnesoid X receptor (FXR) and Takeda G-protein-coupled receptor 5 (TGR5). These two receptors maintain insulin sensitivity and glucose tolerance in both liver and intestine [62][63][29,30]. In addition, other mechanisms have been proposed to account for the increased microbiota capacity to extract energy from the diet intake [64][155]:
-
GM influences energy homeostasis by regulating gene expression via complex mechanisms started by SCFAs and monosaccharides [65][156]. In particular, the commensal microorganisms stimulate monosaccharide cellular uptake [66][157] and induce lipogenesis by activating the transcription factors carbohydrate response element binding protein (ChREBP) and sterol response element binding protein (SREBP) [64][155]. Triacylglycerols, produced trought hepatic lipogenesis, are thus sent from the liver to the blood in the form of very low-density lipoprotein and chylomicrons.
-
HF diet triggers an increased absorption of bacterial lipopolysaccharide (LPS) (an endotoxin in the cell wall of Gram-negative bacteria) from the gut lumen to the bloodstream inducing low-grade inflammation, by activating B cells or dendritic cells activating and cytokine production [67][158]. The inflammation could also be stimulated by endotoxemia condition [66][157]; moreover, also the damaged gut barrier might contribute to this metabolic endotoxaemia [68][159].
Of note, relevant evidence suggests that both the consumption of fermentable carbohydrates and the supplementation of SCFAs result in positive effects on host physiology and energy homeostasis. The resistant starch (RS) is a fermentable dietary fiber used as a carbohydrate source in food. Obanda and colleagues used obesity-prone and obesity-resistant rats to examine how weight gain and fat accretion relate to fermentation levels and GM after feeding RS [69][160]. Obese-prone rats fed with RS at 20% of the weight of the diet did not gain more body fat than the same type of rats fed the same diet except without the RS [69][160]. It could be possible that fermentation of prebiotics increased energy expenditure but with contemporary greater energy absorption, and so without net gain in weight and body fat. The authors hypothesized that dietary RS decreases body fat accumulation through stimulating endogenous GLP-1 and PYY production [70][161]. However, the majority of these recent researches have investigated the effect of SCFAs on animal models or in particular tissue or metabolic process. Since SCFAs have different and parallel metabolic processes that affect energy homeostasis, more studies are needed to bring these effects together in order to elucidate the real impact of SCFAs [46][139].
3.2. Microbiome and the Brain
As previously reported, the gut microbiome affects the host’s CNS functions (as cognitive and vegetative activities) through the MGB axis. CNS functions, vice versa, may influence the structure of the microbiota that inhabits the intestinal lumen [18][112]. Several studies suggest that this mutual interplay has a pivotal role in the occurrence of metabolic disorders, such as diabetes and obesity [71][162], but also in the development of eating and stress-related neuropsychiatric disorders, including [72][163] anxiety and depression.
Currently, researchers are focusing on whether the microbiome can have effects on the CNS process and on the hedonic and homeostatic control of dietary intake [73][164] (Figure 1).
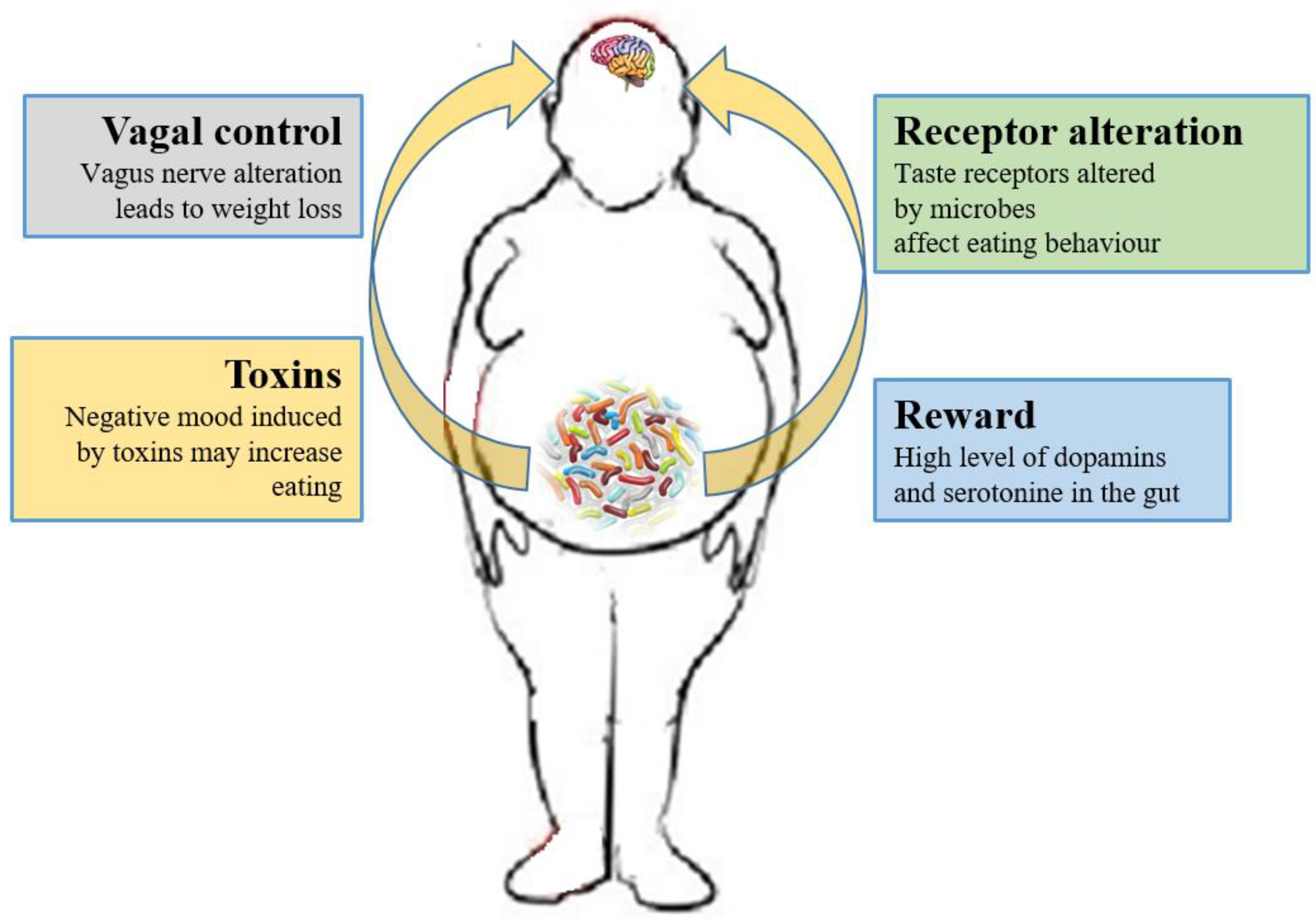
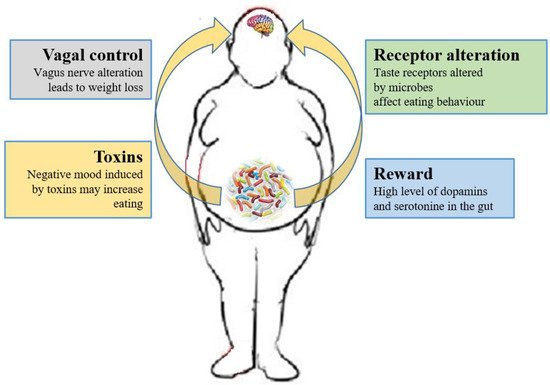
Figure 1. Relations between gut microbiota and eating behavior. The gut microbiota controls the eating behavior by several mechanisms, including changes to receptors such as taste receptors, regulation of reward pathways, production of toxins that alter mood, and deviating neurotransmission via the vagus nerve.
As previously described, the SCFAs produced by the microbiota have systemic effects, but they can also directly signal to the (enteral and central) nervous system, via stimulation of the vagus nerve, or indirectly through immune-neuroendocrine processes [67][158].
The role of the vagus nerve is important in the MGB axis because it connects the 100 million neurons of the enteric nervous system to the “nucleus tractus solitaries” [18][112]. The information inserted in this communication axis is then delivered to the hypothalamus, which modulates energy balance, appetite and dietary intake [74][20]. This information also includes signals from commensal microorganisms, linking the cognitive and emotional nucleus of the CNS with peripheral gut activity, finally leading to host eating control. Experiments showed that transection or blockade of the vagus nerve could induce a dramatic weight loss [75][165]. On the other hand, vagus nerve functions appear to drive extreme eating behavior in satiated animals when they are treated with norepinephrine [76][166]. The parasympathetic vagal activity was linked with weight loss also in anorexia nervosa [77][167], indicating that vagal signaling, involved in the modulation of body weight, can lead to pathological anorexia, and other CNS disorders as anxiety-depressive behaviors and autism [78][79][168,169].
The vagus nerve could be stimulated by enteroendocrine cell hormones as gut peptide YY (PYY) and glucagon-like peptide 1 (GLP-1). The satiety hormone PYY inhibits gut motility, increases gut transit time, and reduces appetite [80][170], while GLP1 decreases appetite and improves insulin sensitivity [81][171]. The bacterial SCFAs could alter the release of those hormones into systemic circulation binding to their specific enteroendocrine G-protein coupled receptors (GPRs) [82][83][172,173]. Through the GPRs activation, SCFAs induce leptin expression, which produces the suppression of the appetite and GLP-1 production [84][174]. The increased plasma GLP-1 and PYY levels inhibit ghrelin secretion [84][174] and regulate appetite by releasing it into the blood stream [85][175]. Acetate, the main SFCA produced by the microbiome, has a direct effect in the suppressing of appetite via central hypothalamic process [59][152]. The increased acetate production, due to an altered gut microbiome, induces the stimulation of the parasympathetic nervous system with improved secretion of ghrelin, obesity and hyperphagia [52][145]. Lactate, another bacterial metabolite produced by Enterobacteriaceae, Lactobacilli and Bifidobacteria, is the favorite substrate for neuronal cells and could prolong the postprandial satiety [86][176].
Furthermore, the microbiome can affect the central control of appetite by producing neuroactive metabolites as tryptophan, serotonin, gamma-aminobutyric acid, endocannabinoid ligands, and ghrelin. These bacterial metabolites are the exact analogs of the mammalian hormones implicated in behavior and mood signaling [87][177]. More than half of the dopamine and the majority of the body’s serotonin are produced at gut level [88][178]. Indeed, components of bacterial flora, as Bacillus cereus, Escherichia coli [89][179], B. subtilis, B. mycoides, Serratia marcescens, Proteus vulgaris, and Staphylococcus aureus [90][180] can produce dopamine. The probiotic B. infantis 35624 improves blood levels of tryptophan [91][181], a precursor of serotonin that mediates appetite-suppressant function by the regulation of melanocorting neurons, which control body weight homeostasis [92][93][182,183]. Moreover, the lactic acid producing bacteria could secrete the neurochemicals histamine [94][184] and GABA [95][185] that is involved in the regulation of feeding and energy balance [96][97][186,187]. Interestingly, GABA stimulates the same neuroreceptors that are targeted by anti-anxiety drugs (benzodiazepines).
3.3. The Role of Microbiome-Driven Inflammation
As widely discussed over the review, the immune system plays a crucial role in the gut–brain axis communications since immune mediators are important messengers of this complex dialogue and, consequently, it mechanistically links the function’s impairments in both brain and gut, as shown by the association between chronic gut inflammation and psychological morbidity [98][99][100][101][211,212,213,214]. The immune system plays a key role in obesity and correlated pathologies, such as in the colorectal cancer [102][215]. In obese patients, a chronic low-grade inflammatory state is maintained [103][104][216,217] and the peripheral inflammation, with the activation of innate immune components (like TLRs) and the loss of intestinal barrier integrity, can lead to neuro-inflammation. Interestingly, recent studies have demonstrated that dysbiosis and inflammation may concur to the development of various diseases, including obesity and depression disorders [105][218]. In addition, numerous studies have now clearly confirmed that the gut microbiome can, qualitatively and quantitatively, shape the host immune responses, both in the gut and in systemic tissues. In this way, the GM influences the concentration and profile of cytokines present in any given individual and, in turn, differentially affects the brain function. For example, GF mice show numerous immune abnormalities, including impaired antibody responses, diminished numbers of T and B lymphocytes and a defective production of cytokines (such as IL-10, TNF-alfa, IL-6 and IL-1) [106][107][219,220]. Moreover, selective GM constituents shape specific aspects of adaptive and innate immunity, including the differentiation of particular effector T-cell lineages [108][109][110][221,222,223]. The obesity-associated dysbiosis is characterized by a remarkable inflammatory potential of microbiota [111][112][224,225], which is able to activate innate and adaptive immunity in the gut and beyond, increasing the inflammatory tone by TLRs activation and production of pro-inflammatory cytokines [113][226]. Sen and colleagues have demonstrated that a dysbiotic microbiota (high sugar diet-associated) alters the vagal gut–brain communication [114][208], producing an inflammatory state that increases gut permeability. The result is the passage of LPS and pro-inflammatory cytokines from the lumen to the lamina propria (triggering an inflammatory response) and so, microglia activation in the nodose ganglion and finally leading to vagal remodeling [111][224]. Moreover, a microbiota with enhanced pro-inflammatory activity has been demonstrated to be able to promote intestinal inflammation, inducing colitis and metabolic syndrome [35][113][14,226]. The loss of intestinal barrier integrity, seems to be a crucial step in the obesity pathogenesis and related diseases, including neurological disorders [106][115][219,227] (Figure 2). In fact, the leaky gut allows the translocation of Gram-negative bacteria’s components into the mesenteric lymph nodes and the circulation, boosting the release of pro-inflammatory cytokines (especially TNF-alpha), via TLR2/4 direct or indirect activation [115][116][117][118][227,228,229,230], and increasing the production of IgA and IgM [119][120][231,232]. In general the gut permeability can be considered the direct consequence of the dysbiotic microbiota-driven local gastrointestinal inflammation [121][122][233,234], and notably, in obese mice, the prebiotics’ supplementation can improve the gut integrity, reducing the weight gain [123][235]. The leaky gut and the associated-inflammation lead to peripheral insulin resistance and hyperglycemia, supporting the obesity establishment; moreover, the increased inflammatory cytokines in the peripheral system can affect the BBB integrity, contributing to the development of mood disorders [122][234]. Bruce-Keller and colleagues have linked obesity, microbiome, and neurologic dysfunction, demonstrating the ability of HF diet-dysbiotic microbiota to increase inflammatory gene expression in the medial prefrontal cortex associated with anxiety and memory impairment [124][236]. Moreover, the inflammation generated by HF diet-dysbiotic microbiota can activate the microglia [125][237], a process observed in various neurological disorders [125][126][127][128][237,238,239,240] and associated with weight gain and bacteria-driven hyperphagia [129][241].
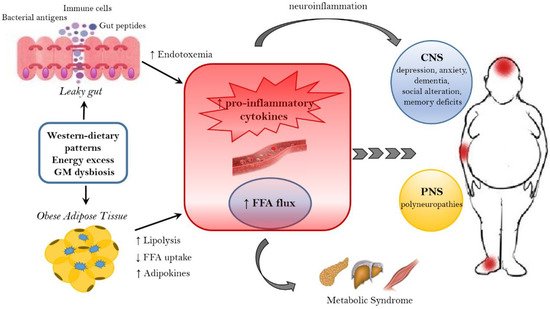
Figure 2. Mechanisms linking obesity to neurological comorbidities. Western-dietary patterns, rich in saturated fat and simple sugars, excessive food intake, and gut microbiota (GM) dysbiosis are related to obesity and its neurological comorbidities through the establishment of an inflammatory state. A dysbiotic microbiota contributes to the leaky gut syndrome, allowing the translocation of gut peptides and bacterial products that increase the peripheral inflammatory tone inducing neuroinflammation. In addition, the dysfunctional obese adipose tissue lead to the increased circulation of inflammatory cytokines, adipokines and FFA. FFA, beside the action on peripheral tissue, where they contribute to the establishment of a metabolic syndrome, have a detrimental effect on both the CNS and PNS. In the CNS, neuroinflammation and lipotoxic FFA can lead to dementia, cognitive impairment, anxiety, and depression, whereas in the PNS the end result are peripheral neuropathies. FFA = free fatty acids. CNS = Central nervous system. PNS = peripheral nervous system.