Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Camila Xu and Version 3 by Camila Xu.
Peroxisome proliferator-activated receptor α is a potent regulator of systemic and cellular metabolism and energy homeostasis, but it also suppresses various inflammatory reactions.
- pattern-recognition receptors
- phagocytosis
- nitric oxide synthase
1. Introduction
Innate immunity comprises a sophisticated set of defensive processes, which are evolutionarily very old and originated concomitantly with the development of multicellular organisms. The defense against invading pathogens is a crucial physiological mechanism that guarantees survival. The development of these mechanisms is a manifestation of a constant race between pathogens (including unicellular pro- and eukaryotic invaders) and host. The biological processes involved in the innate immune response are very complex and tightly regulated on multiple levels, because they may be very harmful when left unsupervised. Recent advances in the elucidation of such a regulation revealed a dense network of connections among immune cell functions, signaling pathways, and cellular metabolism.
2. Peroxisome Proliferator-Activated Receptor alpha (PPARα) and Its Role in Inflammation
Tissue injury and the onset of infection immediately evoke an innate immune response and trigger inflammation. As pointed out by Roman scholar Aulus Cornelius Celsus in the first century, local acute inflammation is manifested by calor, rubor, dolor, and tumor, i.e., increased temperature, redness, pain, and edema [1]. These symptoms reflect the action of proinflammatory lipid mediators, histamine, and cytokines released by tissue-infiltrating leukocytes that induce vasodilation and increase endothelial permeability and expression of adhesion molecules on the endothelial surface and in the extracellular matrix underneath. These events lead to extravasation of circulation leukocytes, chemotaxis, and accumulation of interstitial fluid, causing edema (tumor). The increased interstitial flow and metabolic activity of proliferating cells generate local heat and flushing (calor and rubor). Inflammatory pain (dolor) is evoked by activation of transient receptor potential cation channel vanilloid subfamily member 1 (TRPV1), which is present on sensory neurons of the peripheral nervous system [2]. The TRPV1 activation leads to an influx of Ca2+ and membrane depolarization, followed by the opening of voltage-gated sodium channels and creation of an action potential [2]. TRPV1 receptors are present not only on neurons, but also on immunocompetent cells (T lymphocytes, mast cells), epithelia, keratinocytes, and vascular endothelial cells [3]. TRPV1 channels are activated by various lipid inflammatory mediators, such as COX-2 products (prostaglandins), lipoxygenase 15-LOX products (e.g., 15-hydroperoxyeicosatetraenoic acid, 15-HPETE), and polyamines of molecules released after cell injury, e.g., ATP and adenosine [2]. The links between PPARα and molecular events that spark inflammation and underlie its main symptoms are outlined below (Figure 1).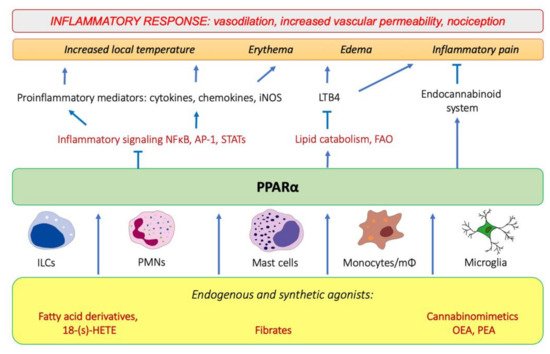
Figure 1. The involvement of PPARα in the modulation of inflammation through interfering with the main inflammatory transcription factors (NF-κB, nuclear factor κB; AP-1, activation protein 1; STATs, signal transducers and activators of transcription) through activating lipid catabolic pathways and participating in the endocannabinoid system (see Section 7). iNOS, inducible nitric oxide synthase; FAO, fatty-acid oxidation; LTB4, leukotriene B4; OEA, oleylethanolamide; PEA, palmitoylethanolamide.
2.1. PPARα as a Nuclear Receptor Present in Peripheral Tissues and Immunocompetent Cells
Peroxisome proliferator-activated receptors (PPARs) belong to a family of nuclear receptors that act as transcription factors activated by lipid-soluble ligands. Such ligands are able to cross the plasma membrane directly and bind the intracellular target proteins. PPARs are represented by three isotypes, PPARα, PPARβ/δ, and PPARγ, encoded by separate genes. They show tissue-specific expression patterns and mainly govern lipid, carbohydrate, and amino-acid metabolism, as well as exert other pleiotropic functions, including immunomodulatory activities. All three PPAR isotypes exhibit potent anti-inflammatory properties and have a strong impact on various aspects of the physiology of the immune system. In this review, we focus on peroxisome proliferator-activated receptor alpha (PPARα), which is particularly responsible for the regulation of fatty-acid catabolism and ketogenesis [4][5], also in addition to being deeply involved in the modulation of innate immunity responses. Below, we outline the active participation of PPARα in physiological processes that operate behind all four cardinal symptoms of inflammation, i.e., alleviating edema and pain and contributing to resolution of acute phase.
As a transcription factor, PPARα is involved in the activation of gene transcription, which is carried out by binding the heterodimer of PPARα and the pan-PPAR obligatory partner, retinoid X receptor (RXR), to consensus motifs in the target promoters. The active heterodimer is formed when both partners have their agonists bound. The most potent endogenous PPARα agonists include fatty acids and their derivatives: saturated stearic and palmitic acids, fatty acyl amides such as oleylethanolamide (OEA) and palmitoylethanolamide (PEA), LOX products such as 5-(S)-HETE and 8-(S)-HETE, and leukotriene B4 (LTB4) [6][7][8][9]. There is the only one bona fide RXR ligand known so far, which is 9-cis-13,14-dihydroretinoic acid, successfully identified after many years of searching, whereas 9-cis-retinoic acid, frequently used experimentally, is one of the most potent pharmacological RXR agonists [10][11]. Pharmacological PPARα agonists, such as fibrates, are clinically used to normalize blood lipid profile, particularly to lower concentrations of cholesterol and low-density lipoprotein fractions [12]. Fenofibrate and gemfibrozil are the most widely prescribed drugs from a fibrate group, and they are generally very well tolerated [13]. Nevertheless, some adverse effects have been reported in patients chronically taking fibrates, with myopathy and rhabdomyolysis being the most common problems [14]. The structures of endogenous ligands, as well as the most important synthetic agonists and antagonists, are presented in Table 1.
Table 1. Chemical structures of PPARα endogenous agonists, synthetic agonists used in experimental studies, clinically used pharmacological agonists, and synthetic antagonists, including examples of novel N-phenylsulfonylamide compounds (the structures of 3- and 10- series according to [15]).
| PPARα Agonists and Antagonists | |
|---|---|
| Natural agonists | 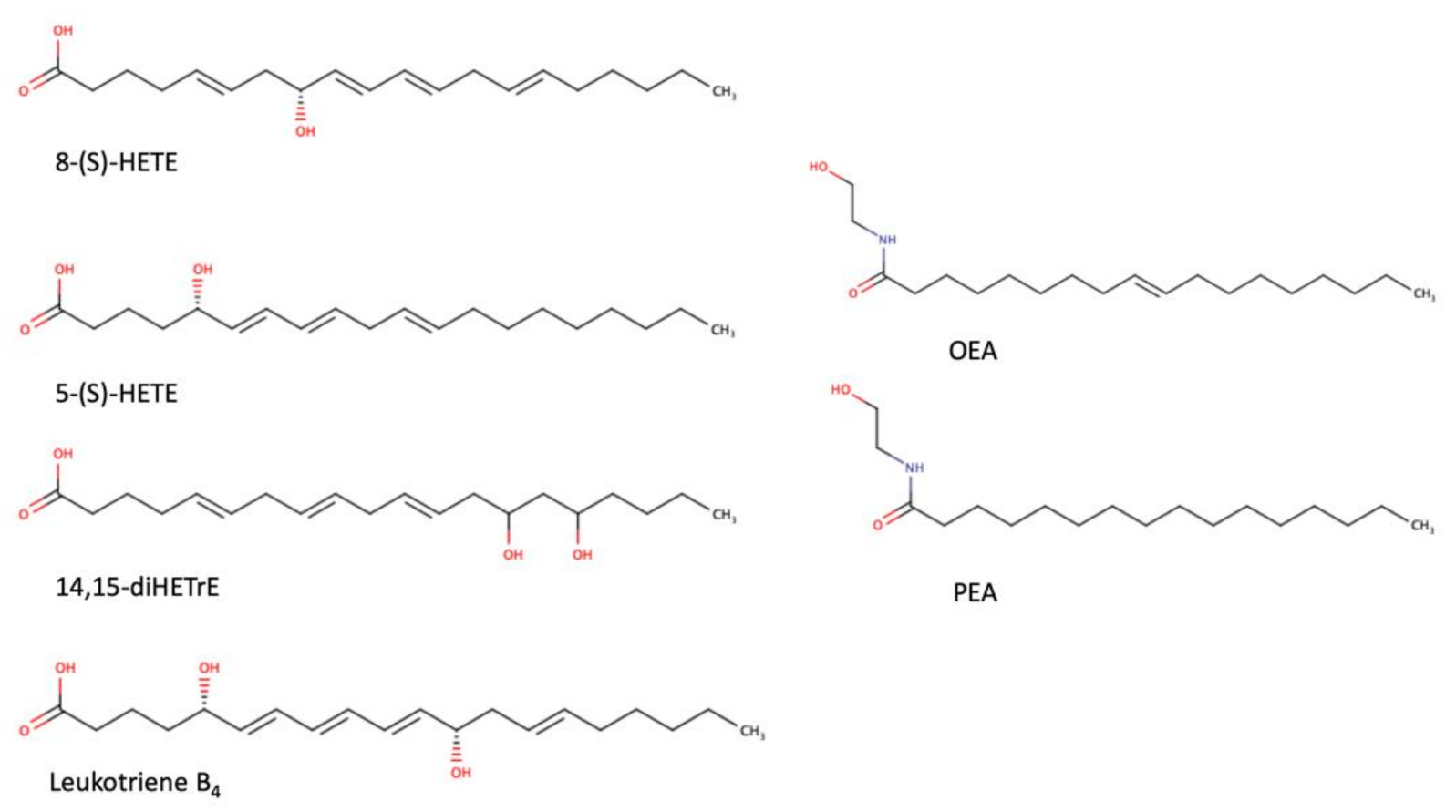 |
| Synthetic agonists | 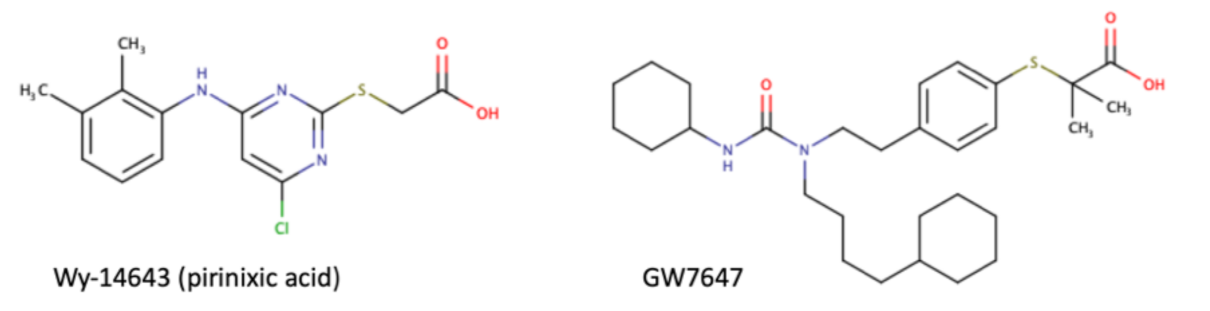 |
| Agonists applied in clinic: fibrate derivatives | 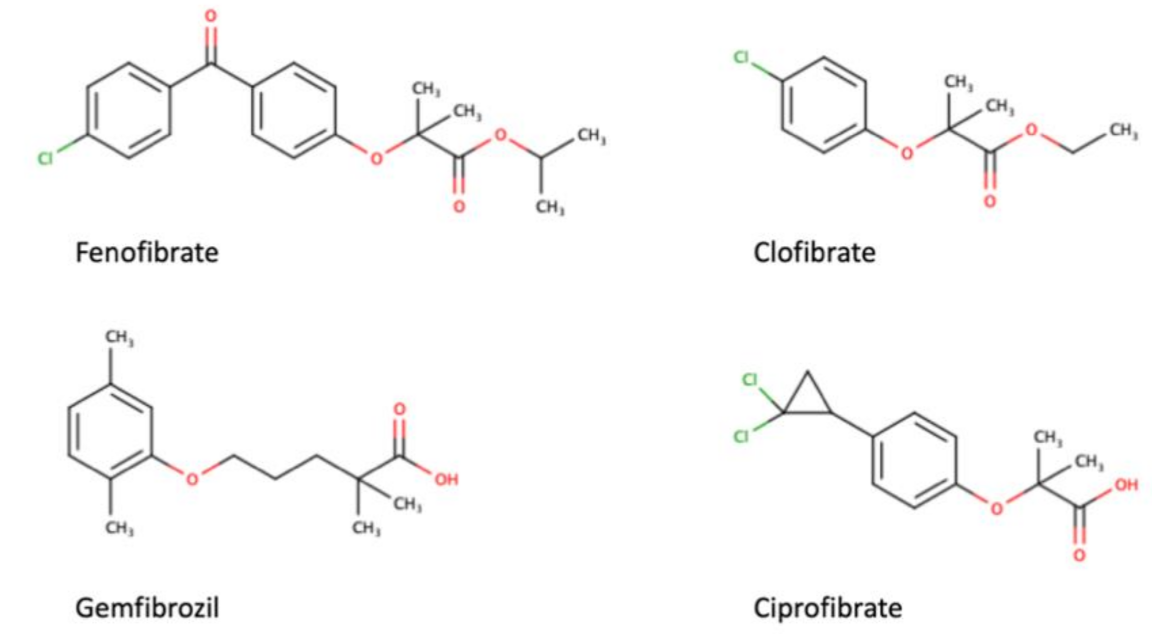 |
| Synthetic antagonists | 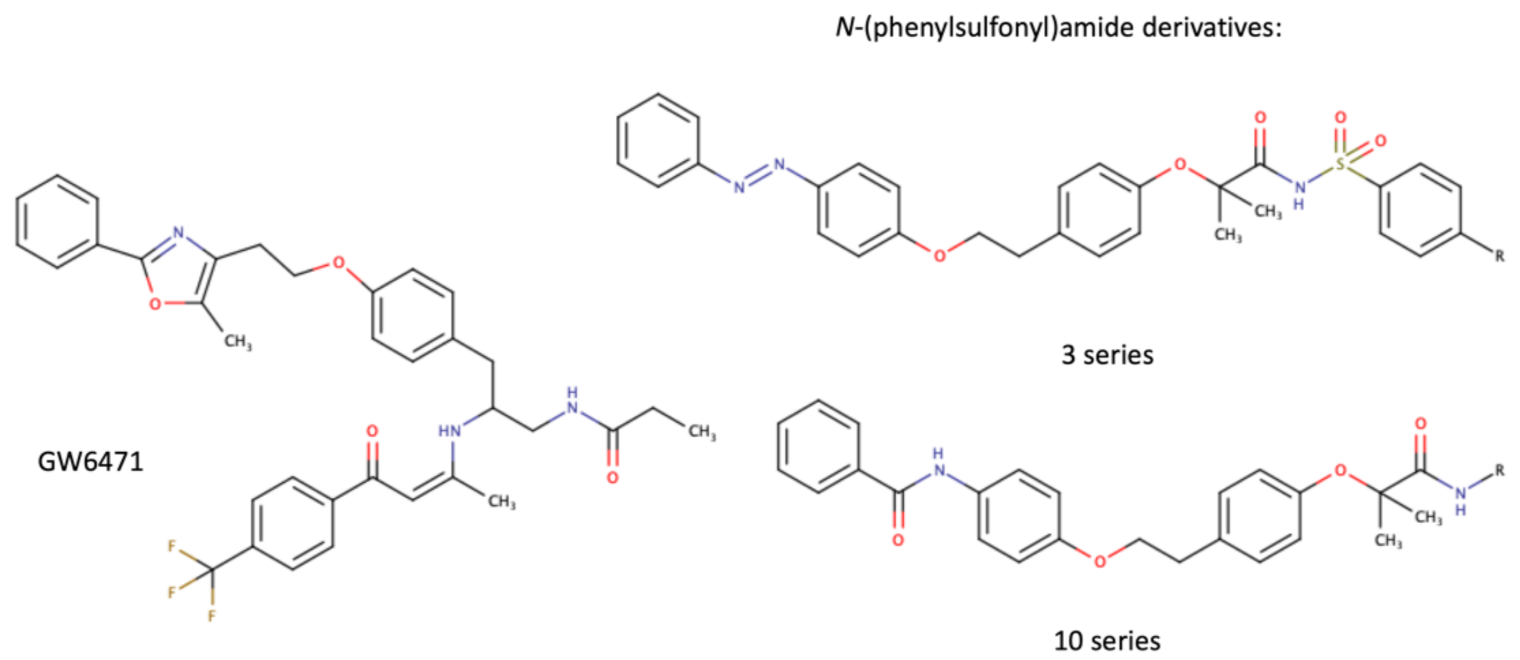 |
Interestingly, in addition to the tissues with a high rate of fatty-acid catabolism, such as the liver, cardiac muscle, and kidneys, PPARα is generally expressed in CD45+ leukocytes [16], including numerous innate immune cell populations: basophils [17], eosinophils [18], monocytes and macrophages [19][20][21][22], Kupffer cells [23], Langerhans cells [24], osteoclasts [25], and microglia [26].
The classical PPARα targets include the genes encoding enzymes from the fatty-acid mitochondrial and peroxisomal β-oxidation (acyl-CoA dehydrogenases, acyl-CoA oxidases), ω-oxidation and ω-hydroxylation (cytochromes P450), and ketogenesis (3-hydroxy-3methylglutaryl-CoA synthase) [27][28][29]. Importantly, in addition to this canonical mode of action, PPARα is able to transrepress certain genes through at least three mechanisms [30]: (i) initiating protein–protein interactions and sequestration of coactivators that are common to PPARα and other pathways, (ii) cross-coupling of the PPARα/RXR complex with other transcription factors, which leads to mutual cross-inhibition of both participating proteins, and (iii) interference with signal-transducing proteins, i.e., where the PPARα/RXR complex inhibits phosphorylation of MAP-kinase cascade members.
2.2. PPARα-Mediated Transrepression of Main Inflammatory Transcription Factors
Transrepressive activity toward nuclear factor κB (NF-κB), activation protein (AP-1), and signal transducers and activators of transcription (STATs) is responsible for PPARα’s profound anti-inflammatory action. PPARα physically interacts with the p65 Rel homology domain through its C-terminal fragment and simultaneously binds the JNK-responsive part of c-Jun with its N-terminal fragment (Figure 2a) [31]. Formation of this complex sequesters p65 and c-Jun from binding to the IL-6 promoter and blocks IL-1-induced IL-6 production. The direct inhibitory interaction between PPARα and NF-κB p65 subunit was also reported in cardiomyocytes [32]. In this case, sirtuin 1 (Sirt1) initiated formation of the Sirt1–PPARα–p65 complex, which led to PPARα-dependent p65 inactivation and transrepression of proinflammatory NF-κB-regulated genes, such as monocyte chemoattractant protein 1 (MCP1, Figure 2b) [32]. Sirt1 induced p65 deacetylation, which also had a negative impact on NF-κB activity because acetylation is required for its activity [33]. The deacetylation effect was absent after treatment with PPARα antagonist GW6471 or in PPARα−/− cells, which indicates PPARα involvement [32].
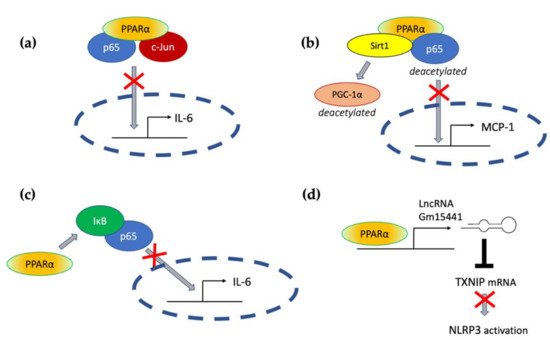
Figure 2. The molecular mechanisms responsible for PPARα-mediated suppression of proinflammatory signaling pathways (see the main text for explanation) (a) through a direct interaction with p65 and c-Jun, (b) through interaction with Sirt1 and subsequent deacetylation of p65, (c) through activation of IκB, and (d) through transactivation of long noncoding RNA Gm15441, which interferes with the stability of thioredoxin-interacting protein (TXNIP) mRNA and blocks NLRP3 inflammasome activation.
An additional mechanism responsible for PPARα interference with the NF-κB pathway was also identified in hepatocytes, where PPARα bound and transactivated NF-κB inhibitor alpha (IκBα), which increased the amount of this protein [34]. Accumulated IκBα binds NF-κB, thereby masking its nuclear localization signal, which arrests it in the cytoplasm and blocks its activity as a transcription factor [35]. PPARα was also responsible for the decreased phosphorylation of NF-κB subunits p65 and p50 [34], which was another event with a negative impact on NF-κB activity, because phosphorylation of its subunits is necessary for their optimal function [36]. The interference of PPARα with NF-κΒ action prevented IL-1 induced IL-6 expression in liver tissues (Figure 2c) [34].
The antagonism between PPARα and NF-κB and AP-1 underlies blocking of the expression of proinflammatory cytokines and effector proteins in various cell and animal models. PPARα ligand K-111 (2,2-dichloro-12-(4-chlorophenyl)-dodecanoic acid) inhibited LPS-induced IL-6 production in Raw 264.7 macrophages on the mRNA and protein level [37]. This effect was exerted through the inhibition of stress-activated protein kinase (SAPK)/c-Jun N-terminal kinase (JNK), NF-κB p65 phosphorylation, and induction of IκBα protein level [37]. PPARα activation in monocytes was shown to inhibit LPS- or IL-1β-induced expression of tissue factor (TF), a membrane glycoprotein responsible for initiation of coagulation cascade [38][39]. The mechanism involved a previously mentioned blockade of the target gene promoter activity through the antagonism between PPARα and NFκB and AP-1 [38].
Interleukins released by immune cells exert their biological functions through specific cell surface receptors, which transduce signals through the Janus family of kinases (JAK) and phosphorylation STAT transcription factors [40]. Various STAT proteins are negatively regulated by PPARα. For example, a bidirectional cross-inhibitory relationship between PPARα and STAT5b was described [41][42][43]. STAT5b is responsible for signal transduction from the IL-2 receptor [44]. IL-2 is a very important cytokine, crucial for both innate and adaptive immunity, being indispensable for NK cell proliferation and maturation, as well as promoting the development, differentiation, and proinflammatory response of both Th1 and Th2 cells [44][45].
2.3. PPARα and Inflammatory Lipid Mediators
Another important mechanism of the anti-inflammatory action of PPARα involves the catabolism of lipid mediators, such as leukotriene B4 (LTB4). The elegant study by Devchand and colleagues [46] revealed that LTB4 is a potent and specific PPARα ligand that induces expression of PPARα-transactivated genes of the peroxisomal β-oxidation pathway, namely, acyl-CoA oxidase, which is a rate-limiting enzyme of LTB4 catabolism. PPARα−/− mice subjected to a topical application of 5-LOX-inducing inflammatory agent and LTB4 showed signs of tissue inflammation much longer (by about 30–40%) than wt mice, which were able to clear LTB4 from circulation much faster [46]. This experiment illustrates the importance of PPARα in the resolution of inflammation. This role of PPARα is necessary for regulation of the innate immune response, because proinflammatory lipid mediators, such as LTB4, are not only strong chemotactic agents for neutrophils and other leukocytes, but they also facilitate PMNs extravasation and diapedesis at the local site of inflammation and increase vascular permeability in this region [47][48]. By restricting LTB4 duration, PPARα alleviates three out of four inflammation symptoms (heat, flushing, and edema). Moreover, PMNs are not only recipients of LTB4 signals, but they are also activated to its production via a positive autocrine feedback loop [49]. Therefore, the PPARα-regulated LTB4 clearance protects from an overexaggerated inflammatory response and its transition from acute to destructive chronic state. The other eicosanoids, the products of either COX, i.e., prostaglandins PGD1, PGD2, PGA1, and PGA2, or 5-LOX product 8-(S)-HETE, also activate PPARα [50], which opens the possibility of modulating their impact on the cells with PPARα expression, whether in immunocompetent cells, such as monocytes/macrophages that express high levels of this receptor, or in the inflamed tissue. Such an activity contributes to tissue protection from inflammatory damage and facilitates regeneration.
2.4. PPARα Crosstalk with Pattern Recognition Receptors
Vertebrates take advantage of the PRR functions and employ them to sense all sorts of factors that induce tissue homeostatic imbalance. The PRR receptors are activated by the numerous compounds comprising specific structural entities referred to as the microbial-associated molecular patterns (MAMPs) or the Damage-associated molecular patterns (DAMPs). Several types of PRRs are broadly present in both immune and nonimmune cells, and their activation sparked by contacts with microorganisms, viruses, and some fragments of damaged cells or an alteration in the functioning of cell components (e.g., cytoskeleton or mitochondria malfunction or endoplasmic reticular stress) is the main trigger of the innate immunity response [51]. The PRRs can be divided into four main subfamilies: the Toll-like receptors (TLRs), the nucleotide-binding oligomerization domain (NOD)–leucin-rich repeat (LRR)-containing receptors (NLRs), the retinoic acid-inducible gene 1-like receptors (RLRs), and the C-type lectin receptors (CLRs) [52]. Nevertheless, some other cellular proteins can serve as PRRs in certain situations, e.g., the glycolytic enzyme, hexokinase II, which is able to spot the microbial sugar, N-acetylglucosamine, when this building block of peptidoglycan happens to be present in the cytoplasm [53]. In this section, we address the question of how PPARα may be involved in the MAMP and DAMP recognition process in various tissues and cells.
The noteworthy information on TLR and PPARα crosstalk comes from the studies on PPARα knockout (KO) mice and cells derived from these animals. The colonic macrophages from KO mice did not produce the regulatory IL-10, but secreted IL-6, IL-1β, and IL-12, potent inducers of Th1 and Th17 differentiation. Moreover, innate immune ILC3 cells isolated from the colon of PPARα KO mice produce lower levels of IL-22 compared with those from WT mice, which results in the impaired secretion of antimicrobial peptides and commensal dysbiosis. This indicates that PPARα regulates the ILC3 effector functions, which are important for both fighting infections and sustaining tolerance to commensal microbiota. The absence of PPARα affects the species composition of the microbiome and leads to increased representation of segmented filamentous bacteria (SFB). All these facts render the KO mice prone to gut inflammation development and are indirect proof of the critical role of PPARα activation in gut immunological homeostasis [19].
It is well known that interactions between the microbiota and intestinal cells engage Toll-like receptors [54], e.g., SFB regulate the process of Th17 differentiation in the intestine via activation of TLR5 by flagellin [55], and TLR4 ligand LPS from Gram-negative bacteria stimulates Th17 differentiation in vitro [56]. It seems that these events can be modulated by PPARα ligands. Accordingly, it was shown that macrophages from PPARα knockout mice are characterized by higher expression levels of mRNA for proinflammatory cytokines IL1β and IL6, as well as for COX-2 and NF-κB (p65) upon TLR4 ligand stimulation (LPS 50 ng/mL, 5 h), as compared to wild-type cells. It seems that PPARα deficiency speeds up LPS-induced inflammatory responses in murine macrophages [21]. Another study on PPARα KO mice indicated that PPARα was essential for the anti-inflammatory effect of acute exercises. Its absence induced overexpression of proinflammatory cytokines in LPS-treated macrophages isolated from mice 24 h post exercise [57].
TLR ligands can regulate PPARα activity, and PPARα agonists influence the expression of TLRs, as well as proteins involved in signaling from TLRs in various cells of both immune and nonimmune types. Becker et al. studied the involvement of LPS in the regulation of PPARα in murine lungs and showed that 24 h on from a prolonged LPS challenge (daily intranasal administration of 1 μg LPS for 4 consecutive days), a profound inhibition of PPARα mRNA expression took place [58]. LPS, peptidoglycan, and flagellin (ligands of TLR4, TLR1/2, and TLR5, respectively) strongly suppressed PPARα activity in rat astrocytes acting at the mRNA and protein expression level [59]. On the other hand, it was shown that fenofibrate, a pharmacological PPAR agonist, significantly inhibited the TLR4, MYD-88, and NF-κB mRNA expression, as well as TNFα production, in murine melanoma B16F10 LPS-stimulated cells [60]. The strong relationship between TLR4 and the PPARα signaling pathway was also clearly demonstrated in a model of endotoxin-induced uveitis. This study suggested that fenofibrate can also attenuate LPS-induced cytokine production, inhibit NF-κB signaling, and suppress TLR4 expression in retinal pigment epithelial cells. Simultaneously, LPS could act as a direct PPARα antagonist in a PPARα reporter cell line [61]. All these experimental data point to a subtle tuning and complicated interplay between activation of PPARα and the TLR signaling pathway, which is needed for the homeostatic balance between triggering and resolution of the inflammatory response in tissues.
2.5. PPARα and the Regulation of Inflammasomes
The inflammasomes, the complex molecular platforms formed in the cytoplasm (mainly in macrophages, but also in other nonimmune cells, such as endothelial and epithelial cells encountering various DAMPs and MAMPs), are now considered the key element of innate immunity. They are the multiprotein complexes composed of cytoplasmic sensors (mainly NLR family members), adaptive proteins (apoptosis-associated speck-like protein, ASC, or PY-CARD), and effectors (such as cysteine proteinase precursor or pro-caspase-1). In the case of some nonconventional inflammasomes, pro-caspase-1 is substituted by pro-caspase-11 in murine cells and pro-caspase 4/5 in human cells. The complex formation enables the proteolysis of pro-IL1β and pro-IL18 and the release of active cytokines into the cell microenvironment and bloodstream, which drives local or systemic inflammation [62]. Alternatively, the inflammasome formation induces a chain of events leading to pyroptosis—the special type of a programmed cell death connected to an inflammatory state. The molecular mechanisms contributing to inflammasome activity are not yet completely understood, but it is believed that the process of their formation requires two subsequent signals, e.g., LPS binding to TLR4 on the cell membrane as the primary signal and K+ efflux, cytosolic release of lysosomal cathepsins, or mitochondria-derived factors and reactive oxygen species generation as the secondary signal [63]. The regulation of inflammasome activation can occur at both signals on the post-transcriptional and post-translational levels [64].
It was shown in some animal models that PPARα activation can profoundly suppress the inflammasome-induced tissue injury, thereby contributing to the resolution of inflammation. This can be partially attributed to the downregulation of TLR expression by PPARα and interference with the primary step of inflammasome activation. However, in PPARα KO mice with lung inflammation caused by Pseudomonas aeruginosa introduction, a significant increase in expression of NLRP-3, ASC-1, and caspase-1, as compared with infected wt mice, was observed [65]. This indicates that PPARα expression background is also important for the supply of inflammasome building blocks.
Acute liver injury is a disease strongly connected with NLPR3 inflammasome activity. In the context of this pathology, Brocker et al. proposed a mechanism connecting fasting, PPARα, and the reduction in liver inflammation and injury. They showed that the long noncoding RNA gene Gm15441 contained a PPARα-binding site within its promoter, and the Gm15441 RNA expression was activated by PPARα ligand Wy-14643. Gm15441 suppressed its antisense transcript, encoding thioredoxin-interacting protein (TXNIP). This subsequently decreased TXNIP-stimulated NLRP3 inflammasome activation (Figure 2d) [66].
Moreover, it was shown that OEA, an endogenous bioactive lipid and a natural ligand of PPARα, prevented tissue damage in the onset of LPS/d-galactosamine (d-Gal)-induced acute liver injury. OEA administration increased PPARα expression in murine liver subjected to LPS/d-Gal treatment. In turn, the liver protein levels of IL-1β and NLRP3 inflammasome components, NLRP3 protein and pro-caspase-1, were enhanced after LPS/d-Gal injection in mice. The increase in these proteins was alleviated by OEA addition to the diet [67]. The OEA anti-inflammatory effects were also evident in dextran sulfate sodium (DSS)-induced mice colitis, and the effect was mediated by the inhibition of NLRP3, NF-κB, or MyD88-dependent pathways [68].
References
- Celsus, A.C. De Medicina, 1st ed.; Nicolaus Laurentii: Florence, Italy, 1478.
- Frias, B.; Merighi, A. Capsaicin, nociception and pain. Molecules 2016, 21, 797.
- Nilius, B.; Owsianik, G. The transient receptor potential family of ion channels. Genome Biol. 2011, 12, 218.
- Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptors: Nuclear control of metabolism. Endocrinol. Rev. 1999, 20, 649–688.
- Sertznig, P.; Seifert, M.; Tilgen, W.; Reichrath, J. Present concepts and future outlook: Function of peroxisome proliferator-activated receptors (PPARs) for pathogenesis, progression, and therapy of cancer. J. Cell Physiol. 2007, 212, 1–12.
- Kamata, S.; Oyama, T.; Saito, K.; Honda, A.; Yamamoto, Y.; Suda, K.; Ishikawa, R.; Itoh, T.; Watanabe, Y.; Shibata, T.; et al. PPARalpha ligand-binding domain structures with endogenous fatty acids and fibrates. iScience 2020, 23, 101727.
- Wahli, W.; Michalik, L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol. Metab. 2012, 23, 351–363.
- Lo Verme, J.; Fu, J.; Astarita, G.; La Rana, G.; Russo, R.; Calignano, A.; Piomelli, D. The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethanolamide. Mol. Pharmacol. 2005, 67, 15–19.
- LoVerme, J.; La Rana, G.; Russo, R.; Calignano, A.; Piomelli, D. The search for the palmitoylethanolamide receptor. Life Sci. 2005, 77, 1685–1698.
- de Lera, A.R.; Krezel, W.; Ruhl, R. An endogenous mammalian retinoid X receptor ligand, at last! ChemMedChem 2016, 11, 1027–1037.
- Ruhl, R.; Krzyzosiak, A.; Niewiadomska-Cimicka, A.; Rochel, N.; Szeles, L.; Vaz, B.; Wietrzych-Schindler, M.; Alvarez, S.; Szklenar, M.; Nagy, L.; et al. 9-cis-13,14-dihydroretinoic acid is an endogenous retinoid acting as RXR ligand in mice. PLoS Genet. 2015, 11, e1005213.
- Najib, J. Fenofibrate in the treatment of dyslipidemia: A review of the data as they relate to the new suprabioavailable tablet formulation. Clin. Ther. 2002, 24, 2022–2050.
- Blais, J.E.; Tong, G.K.Y.; Pathadka, S.; Mok, M.; Wong, I.C.K.; Chan, E.W. Comparative efficacy and safety of statin and fibrate monotherapy: A systematic review and meta-analysis of head-to-head randomized controlled trials. PLoS ONE 2021, 16, e0246480.
- Alsheikh-Ali, A.A.; Kuvin, J.T.; Karas, R.H. Risk of adverse events with fibrates. Am. J. Cardiol. 2004, 94, 935–938.
- Ammazzalorso, A.; Bruno, I.; Florio, R.; De Lellis, L.; Laghezza, A.; Cerchia, C.; De Filippis, B.; Fantacuzzi, M.; Giampietro, L.; Maccallini, C.; et al. Sulfonimide and amide derivatives as novel PPARalpha antagonists: Synthesis, antiproliferative activity, and docking studies. ACS Med. Chem. Lett. 2020, 11, 624–632.
- Kaipainen, A.; Kieran, M.W.; Huang, S.; Butterfield, C.; Bielenberg, D.; Mostoslavsky, G.; Mulligan, R.; Folkman, J.; Panigrahy, D. PPARalpha deficiency in inflammatory cells suppresses tumor growth. PLoS ONE 2007, 2, e260.
- Fujimura, Y.; Tachibana, H.; Yamada, K. Peroxisome proliferator-activated receptor ligands negatively regulate the expression of the high-affinity IgE receptor Fc epsilon RI in human basophilic KU812 cells. Biochem. Biophys. Res. Commun. 2002, 297, 193–201.
- Woerly, G.; Honda, K.; Loyens, M.; Papin, J.P.; Auwerx, J.; Staels, B.; Capron, M.; Dombrowicz, D. Peroxisome proliferator-activated receptors alpha and gamma down-regulate allergic inflammation and eosinophil activation. J. Exp. Med. 2003, 198, 411–421.
- Manoharan, I.; Suryawanshi, A.; Hong, Y.; Ranganathan, P.; Shanmugam, A.; Ahmad, S.; Swafford, D.; Manicassamy, B.; Ramesh, G.; Koni, P.A.; et al. Homeostatic PPARalpha signaling limits inflammatory responses to commensal microbiota in the intestine. J. Immunol. 2016, 196, 4739–4749.
- Chinetti, G.; Fruchart, J.C.; Staels, B. Peroxisome proliferator-activated receptors: New targets for the pharmacological modulation of macrophage gene expression and function. Curr. Opin. Lipidol. 2003, 14, 459–468.
- Babaev, V.R.; Ishiguro, H.; Ding, L.; Yancey, P.G.; Dove, D.E.; Kovacs, W.J.; Semenkovich, C.F.; Fazio, S.; Linton, M.F. Macrophage expression of peroxisome proliferator-activated receptor-alpha reduces atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation 2007, 116, 1404–1412.
- Wu, L.; Zhang, X.; Zheng, L.; Zhao, H.; Yan, G.; Zhang, Q.; Zhou, Y.; Lei, J.; Zhang, J.; Wang, J.; et al. RIPK3 orchestrates fatty acid metabolism in tumor-associated macrophages and hepatocarcinogenesis. Cancer Immunol. Res. 2020, 8, 710–721.
- Brocker, C.N.; Yue, J.; Kim, D.; Qu, A.; Bonzo, J.A.; Gonzalez, F.J. Hepatocyte-specific PPARA expression exclusively promotes agonist-induced cell proliferation without influence from nonparenchymal cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G283–G299.
- Dubrac, S.; Stoitzner, P.; Pirkebner, D.; Elentner, A.; Schoonjans, K.; Auwerx, J.; Saeland, S.; Hengster, P.; Fritsch, P.; Romani, N.; et al. Peroxisome proliferator-activated receptor-alpha activation inhibits Langerhans cell function. J. Immunol. 2007, 178, 4362–4372.
- Poulsen, R.C.; Moughan, P.J.; Kruger, M.C. Long-chain polyunsaturated fatty acids and the regulation of bone metabolism. Exp. Biol. Med. 2007, 232, 1275–1288.
- Warden, A.; Truitt, J.; Merriman, M.; Ponomareva, O.; Jameson, K.; Ferguson, L.B.; Mayfield, R.D.; Harris, R.A. Localization of PPAR isotypes in the adult mouse and human brain. Sci. Rep. 2016, 6, 27618.
- Kroetz, D.L.; Yook, P.; Costet, P.; Bianchi, P.; Pineau, T. Peroxisome proliferator-activated receptor alpha controls the hepatic CYP4A induction adaptive response to starvation and diabetes. J. Biol. Chem. 1998, 273, 31581–31589.
- Hashimoto, T.; Fujita, T.; Usuda, N.; Cook, W.; Qi, C.; Peters, J.M.; Gonzalez, F.J.; Yeldandi, A.V.; Rao, M.S.; Reddy, J.K. Peroxisomal and mitochondrial fatty acid beta-oxidation in mice nullizygous for both peroxisome proliferator-activated receptor alpha and peroxisomal fatty acyl-CoA oxidase. Genotype correlation with fatty liver phenotype. J. Biol. Chem. 1999, 274, 19228–19236.
- Vila-Brau, A.; De Sousa-Coelho, A.L.; Mayordomo, C.; Haro, D.; Marrero, P.F. Human HMGCS2 regulates mitochondrial fatty acid oxidation and FGF21 expression in HepG2 cell line. J. Biol. Chem. 2011, 286, 20423–20430.
- Daynes, R.A.; Jones, D.C. Emerging roles of PPARs in inflammation and immunity. Nat. Rev. Immunol. 2002, 2, 748–759.
- Delerive, P.; De Bosscher, K.; Besnard, S.; Vanden Berghe, W.; Peters, J.M.; Gonzalez, F.J.; Fruchart, J.C.; Tedgui, A.; Haegeman, G.; Staels, B. Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappaB and AP-1. J. Biol. Chem. 1999, 274, 32048–32054.
- Planavila, A.; Iglesias, R.; Giralt, M.; Villarroya, F. Sirt1 acts in association with PPARalpha to protect the heart from hypertrophy, metabolic dysregulation, and inflammation. Cardiovasc. Res. 2011, 90, 276–284.
- Rothgiesser, K.M.; Fey, M.; Hottiger, M.O. Acetylation of p65 at lysine 314 is important for late NF-kappaB-dependent gene expression. BMC Genom. 2010, 11, 22.
- Zhang, N.; Chu, E.S.; Zhang, J.; Li, X.; Liang, Q.; Chen, J.; Chen, M.; Teoh, N.; Farrell, G.; Sung, J.J.; et al. Peroxisome proliferator activated receptor alpha inhibits hepatocarcinogenesis through mediating NF-kappaB signaling pathway. Oncotarget 2014, 5, 8330–8340.
- Jacobs, M.D.; Harrison, S.C. Structure of an IkappaBalpha/NF-kappaB complex. Cell 1998, 95, 749–758.
- Chen, L.F.; Greene, W.C. Shaping the nuclear action of NF-kappaB. Nat. Rev. Mol. Cell Biol. 2004, 5, 392–401.
- Murakami, K.; Bujo, H.; Unoki, H.; Saito, Y. Effect of PPARalpha activation of macrophages on the secretion of inflammatory cytokines in cultured adipocytes. Eur. J. Pharmacol. 2007, 561, 206–213.
- Marx, N.; Mackman, N.; Schonbeck, U.; Yilmaz, N.; Hombach, V.; Libby, P.; Plutzky, J. PPARalpha activators inhibit tissue factor expression and activity in human monocytes. Circulation 2001, 103, 213–219.
- Neve, B.P.; Corseaux, D.; Chinetti, G.; Zawadzki, C.; Fruchart, J.C.; Duriez, P.; Staels, B.; Jude, B. PPARalpha agonists inhibit tissue factor expression in human monocytes and macrophages. Circulation 2001, 103, 207–212.
- Haque, S.J.; Sharma, P. Interleukins and STAT signaling. Vitam. Horm. 2006, 74, 165–206.
- Shipley, J.M.; Waxman, D.J. Down-regulation of STAT5b transcriptional activity by ligand-activated peroxisome proliferator-activated receptor (PPAR) alpha and PPARgamma. Mol. Pharmacol. 2003, 64, 355–364.
- Shipley, J.M.; Waxman, D.J. Simultaneous, bidirectional inhibitory crosstalk between PPAR and STAT5b. Toxicol. Appl. Pharmacol. 2004, 199, 275–284.
- Zhou, Y.C.; Waxman, D.J. STAT5b down-regulates peroxisome proliferator-activated receptor alpha transcription by inhibition of ligand-independent activation function region-1 trans-activation domain. J. Biol. Chem. 1999, 274, 29874–29882.
- Lin, J.X.; Leonard, W.J. The role of Stat5a and Stat5b in signaling by IL-2 family cytokines. Oncogene 2000, 19, 2566–2576.
- Bendickova, K.; Fric, J. Roles of IL-2 in bridging adaptive and innate immunity, and as a tool for cellular immunotherapy. J. Leukoc. Biol. 2020, 108, 427–437.
- Devchand, P.R.; Keller, H.; Peters, J.M.; Vazquez, M.; Gonzalez, F.J.; Wahli, W. The PPARalpha-leukotriene B4 pathway to inflammation control. Nature 1996, 384, 39–43.
- Dahlen, S.E.; Bjork, J.; Hedqvist, P.; Arfors, K.E.; Hammarstrom, S.; Lindgren, J.A.; Samuelsson, B. Leukotrienes promote plasma leakage and leukocyte adhesion in postcapillary venules: In vivo effects with relevance to the acute inflammatory response. Proc. Natl. Acad. Sci. USA 1981, 78, 3887–3891.
- Lindbom, L.; Hedqvist, P.; Dahlen, S.E.; Lindgren, J.A.; Arfors, K.E. Leukotriene B4 induces extravasation and migration of polymorphonuclear leukocytes in vivo. Acta Physiol. Scand. 1982, 116, 105–108.
- Marleau, S.; Fruteau de Laclos, B.; Sanchez, A.B.; Poubelle, P.E.; Borgeat, P. Role of 5-lipoxygenase products in the local accumulation of neutrophils in dermal inflammation in the rabbit. J. Immunol. 1999, 163, 3449–3458.
- Yu, K.; Bayona, W.; Kallen, C.B.; Harding, H.P.; Ravera, C.P.; McMahon, G.; Brown, M.; Lazar, M.A. Differential activation of peroxisome proliferator-activated receptors by eicosanoids. J. Biol. Chem. 1995, 270, 23975–23983.
- Christofides, A.; Konstantinidou, E.; Jani, C.; Boussiotis, V.A. The role of peroxisome proliferator-activated receptors (PPAR) in immune responses. Metabolism 2021, 114, 154338.
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol. 2018, 9, 2379.
- Wolf, A.J.; Reyes, C.N.; Liang, W.; Becker, C.; Shimada, K.; Wheeler, M.L.; Cho, H.C.; Popescu, N.I.; Coggeshall, K.M.; Arditi, M.; et al. Hexokinase Is an innate immune receptor for the detection of bacterial peptidoglycan. Cell 2016, 166, 624–636.
- Frosali, S.; Pagliari, D.; Gambassi, G.; Landolfi, R.; Pandolfi, F.; Cianci, R. How the intricate interaction among toll-like receptors, microbiota, and intestinal immunity can influence gastrointestinal pathology. J. Immunol. Res. 2015, 2015, 489821.
- Wang, Y.; Yin, Y.; Chen, X.; Zhao, Y.; Wu, Y.; Li, Y.; Wang, X.; Chen, H.; Xiang, C. Induction of intestinal Th17 cells by flagellins from segmented filamentous bacteria. Front. Immunol. 2019, 10, 2750.
- Park, J.H.; Jeong, S.Y.; Choi, A.J.; Kim, S.J. Lipopolysaccharide directly stimulates Th17 differentiation in vitro modulating phosphorylation of RelB and NF-kappaB1. Immunol. Lett. 2015, 165, 10–19.
- Silveira, L.S.; Pimentel, G.D.; Souza, C.O.; Biondo, L.A.; Teixeira, A.A.S.; Lima, E.A.; Batatinha, H.A.P.; Rosa Neto, J.C.; Lira, F.S. Effect of an acute moderate-exercise session on metabolic and inflammatory profile of PPAR-alpha knockout mice. Cell Biochem. Funct. 2017, 35, 510–517.
- Becker, J.; Delayre-Orthez, C.; Frossard, N.; Pons, F. Regulation of peroxisome proliferator-activated receptor-alpha expression during lung inflammation. Pulm. Pharmacol. Ther. 2008, 21, 324–330.
- Chistyakov, D.V.; Aleshin, S.E.; Astakhova, A.A.; Sergeeva, M.G.; Reiser, G. Regulation of peroxisome proliferator-activated receptors (PPAR) alpha and -gamma of rat brain astrocytes in the course of activation by toll-like receptor agonists. J. Neurochem. 2015, 134, 113–124.
- Dana, N.; Javanmard, H.S.; Vaseghi, G. The effect of fenofibrate, a PPARalpha activator on toll-like receptor-4 signal transduction in melanoma both in vitro and in vivo. Clin. Transl. Oncol. 2020, 22, 486–494.
- Shen, W.; Gao, Y.; Lu, B.; Zhang, Q.; Hu, Y.; Chen, Y. Negatively regulating TLR4/NF-kappaB signaling via PPARalpha in endotoxin-induced uveitis. Biochim. Biophys. Acta 2014, 1842, 1109–1120.
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022.
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome activation and regulation: Toward a better understanding of complex mechanisms. Cell Discov. 2020, 6, 36.
- Seok, J.K.; Kang, H.C.; Cho, Y.Y.; Lee, H.S.; Lee, J.Y. Regulation of the NLRP3 inflammasome by post-translational modifications and small molecules. Front. Immunol. 2020, 11, 618231.
- Gugliandolo, E.; Fusco, R.; Ginestra, G.; D’Amico, R.; Bisignano, C.; Mandalari, G.; Cuzzocrea, S.; Di Paola, R. Involvement of TLR4 and PPAR-alpha receptors in host response and NLRP3 Inflammasome activation, against pulmonary infection with Pseudomonas aeruginosa. Shock 2019, 51, 221–227.
- Brocker, C.N.; Kim, D.; Melia, T.; Karri, K.; Velenosi, T.J.; Takahashi, S.; Aibara, D.; Bonzo, J.A.; Levi, M.; Waxman, D.J.; et al. Long non-coding RNA Gm15441 attenuates hepatic inflammasome activation in response to PPARA agonism and fasting. Nat. Commun. 2020, 11, 5847.
- Hu, J.; Zhu, Z.; Ying, H.; Yao, J.; Ma, H.; Li, L.; Zhao, Y. Oleoylethanolamide protects against acute liver injury by regulating Nrf-2/HO-1 and NLRP3 pathways in mice. Front. Pharmacol. 2020, 11, 605065.
- Lama, A.; Provensi, G.; Amoriello, R.; Pirozzi, C.; Rani, B.; Mollica, M.P.; Raso, G.M.; Ballerini, C.; Meli, R.; Passani, M.B. The anti-inflammatory and immune-modulatory effects of OEA limit DSS-induced colitis in mice. Biomed. Pharmacother. 2020, 129, 110368.
More