Gluten-related disorders (GRDs) are a group of diseases that involve the activation of the immune system triggered by the ingestion of gluten, with a worldwide prevalence of 5%. Among them, Celiac disease (CeD) is a T-cell-mediated autoimmune disease causing a plethora of symptoms from diarrhea and malabsorption to lymphoma. Even though GRDs have been intensively studied, the environmental triggers promoting the diverse reactions to gluten proteins in susceptible individuals remain elusive. It has been proposed that pathogens could act as disease-causing environmental triggers of CeD by molecular mimicry mechanisms. Additionally, it could also be possible that unrecognized molecular, structural, and physical parallels between gluten and bacteria have a relevant role.
- non-celiac gluten sensitivity
- celiac disease
- molecular mimicry
- gliadin peptides
1. Introduction
Celiac disease (CeD) is a chronic, small-intestinal T-cell-mediated autoimmune disease triggered by the ingestion of dietary gluten from common food grains such as wheat, rye, and barley, in genetically predisposed individuals with a prevalence of about 1% in the general population with regional differences [1]. Other gluten-related disorders (GRDs) such as non-celiac gluten sensitivity (NCGS) are less understood and have a prevalence rate between 0.5% and 5% [2]. Nowadays, the only proven treatment for GRDs is strict and life-long adherence to a gluten-free diet [3]. Viral and bacterial pathogens have long been suspected of triggering immune responses that are directed toward autoimmunity in CeD. In 2002, the group of Khosla showed that the immunodominant gluten fragment has sequence similarity with pertactin, a highly immunogenic protein from the bacterium Bordetella pertussis; however, this result was not further investigated [4]. It recently identified and characterized a number of mimics of HLA-DQ2.5-restricted gliadin determinants derived from the commensal bacterium Pseudomonas fluorescens, activating disease-relevant gliadin reactive T cells isolated from CeD patients [5]. This report was a major proof of concept that a molecular mimicry mechanism may trigger CeD. Beyond T-cell activation in CeD, it was also proposed that gluten proteins have functional similarities with non-replicative pathogens such as prions [6][7]. The main issue with gluten proteins is that the human digestive proteases can only partially degrade them leading to a mixture of peptides that elicit immune and toxic effects in predisposed individuals and cell lines [8]. The mononuclear phagocytic system (MPS), composed mainly of macrophages, neutrophils, and dendritic cells, is part of the first line of defense against pathogens. Pathogens are recognized by various immune cells, such as macrophages and dendritic cells, via pathogen-associated molecular patterns (PAMPs) on the pathogen surface, which interact with complementary pattern-recognition receptors (PRRs) on the pathogen surface the immune cell surfaces. While PRR activation is a central component to the resulting immune response, innate cells also respond to the size and shape of the pathogens and the spatial organization of PAMPs on the bacterial surface. In recent years, it has become evident that the MPS can engulf other non-self-systems such as synthetic nanoparticles generating in many cases an immune response against the foreign nanosystem [9][10]. From a pathological standpoint, there are some concerns in the possibility that the presence of nanostructures could exacerbate or prolong PRR-driven inflammatory reactions, leading to uncontrolled tissue-damaging inflammation. In this context, it was demonstrated that pepsin digests of gliadin form spontaneously amyloid-like structures that trigger genes in the gut epithelial cell model Caco-2 involved in recruiting specialized immune cells [11]. Furthermore, the most studied gluten peptides are 33-mer peptides and the p31-43 fragment that form peptide nanostructures, too. Both peptides trigger the adaptive or an innate immune response, respectively, in CeD patients, animal models, and cell lines culture [12][13][14][15][16]. The immunodominant 33-mer peptide comprises residues 57 to 89 of α-2-gliadin (LQLQPFPQPQLPYPQPQLPYPQPQLPYPQPQPF). In total, 39% of the 33-mer peptide residues are prolines leading to a type-II polyproline (PPII) conformation in solution, which is known to be bound to MHC class-II molecules [14]. When the 33-mer accumulates, it forms superstructures ranging from dimers to nano- and microstructures ranging from 10 nm to more than 1 µm [17][18][19]. Importantly, there is a concentration-dependent structural transition from PPII toward a parallel β-sheet conformation accompanied by the formation of large superstructures that activate macrophages in vitro via Toll-like receptor 4 (TLR4) and TLR2 [20][21]. The mentioned secondary structures are essential in protein-protein interaction and might function as a signaling component. The high percentage of Q makes this peptide amphiphilic favoring the formation of hydrogen bonds [22], a key factor in the self-assembling process, stressing the importance between sequence and morphology [17]. The second most studied gliadin peptide is the toxic p-31-43, a 13-mer peptide comprising the amino acids 31 to 43 (FPGQQQPFPPQQP). Although the p31-43 peptide is not presented by the HLA-DQ2 [23], the reason for its toxicity in CeD remains unknown. The mechanism by which p31-43 induces an immune response in celiac patients has been recently attributed to its effects on the endocytic compartment affecting several cell functions such as proliferation, cell motility, and innate immunity activation [23]. Nanayakkara et al. [24] recently proposed that p31-43 induces the IFN-α-mediated innate immune response in the CaCo-2 enterocyte cell-line by activating the TLR7 signaling pathway mimicking the immune response triggered by viruses. Recently, it was reported that p31-43 has a PPII secondary structure and can, as well as 33-mer, self-assemble under physiologically relevant conditions [25]. Even more, p31-43 oligomers have been proposed to be responsible for activating the inflammasome in murine models [26]. In-depth sequence and structural analysis of foreign proteins sharing high sequence similarity regions with the 33-mer and p31-43 sequences found some interesting novel bacteria connections. The top-five BLASTp hits using the 33-mer were S. viridochromogenes, Fischerella sp., Granulicatella sp., S. pneumoniae and Nostoc sp. Figure 1. In the case of the p31-43 sequence, Streptomyces sp. and L. edodes were found Figure 2. Overall the identified similarity regions reached up to 68% sequence identity in the 33-mer and up to 85% for the p31-43 similarity regions. Importantly, 5 out of 10 identified proteins belonging to the host organisms known to be pathogenic for humans to different degrees.2. Primary Structure Analysis and Potential Functions
2.1. CeD-T-Cell Epitopes
2.2. SH3/WW Domains Binders
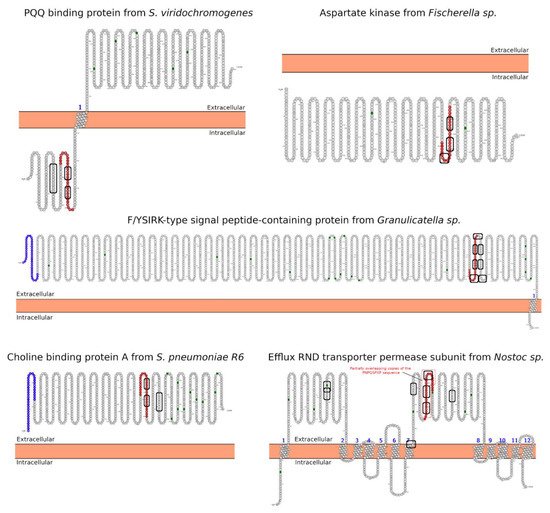
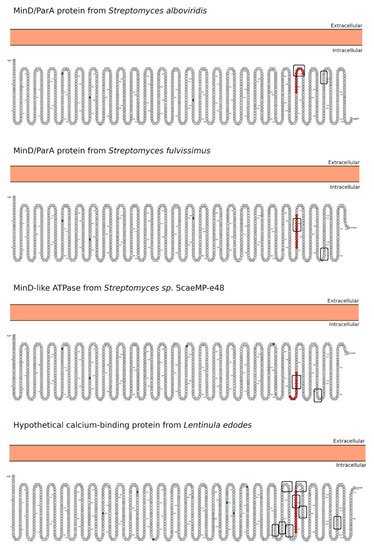
3. Morphology Mimicry
References
- Cabanillas, B. Gluten-related disorders: Celiac disease, wheat allergy, and nonceliac gluten sensitivity. Crit. Rev. Food Sci. Nutr. 2020, 60, 2606–2621.
- Taraghikhah, N.; Ashtari, S.; Asri, N.; Shahbazkhani, B.; Al-Dulaimi, D.; Rostami-Nejad, M.; Rezaei-Tavirani, M.; Razzaghi, M.R.; Zali, M.R. An updated overview of spectrum of gluten-related disorders: Clinical and diagnostic aspects. BMC Gastroenterol. 2020, 20, 258.
- Di Sabatino, A.; Corazza, G.R. Coeliac disease. Lancet 2009, 373, 1480–1493.
- Shan, L.; Molberg, Ø.; Parrot, I.; Hausch, F.; Filiz, F.; Gray, G.M.; Sollid, L.M.; Khosla, C. Structural Basis for Gluten Intolerance in Celiac Sprue. Science 2002, 297, 2275–2279.
- Petersen, J.; Ciacchi, L.; Tran, M.T.; Loh, K.L.; Kooy-Winkelaar, Y.; Croft, N.P.; Hardy, M.I.; Chen, Z.; McCluskey, J.; Anderson, R.P.; et al. T cell receptor cross-reactivity between gliadin and bacterial peptides in celiac disease. Nat. Struct. Mol. Biol. 2020, 27, 49–61.
- Bethune, M.T.; Khosla, C. Parallels between Pathogens and Gluten Peptides in Celiac Sprue. PLoS Pathog. 2008, 4, e34.
- Verdu, E.F.; Schuppan, D. The enemy within the gut: Bacterial pathogens in celiac autoimmunity. Nat. Struct. Mol. Biol. 2019, 27, 5–7.
- Lammers, K.M.; Herrera, M.G.; Dodero, V.I. Translational Chemistry Meets Gluten-Related Disorders. ChemistryOpen 2018, 7, 217–232.
- Doshi, N.; Mitragotri, S. Macrophages Recognize Size and Shape of Their Targets. PLoS ONE 2010, 5, e10051.
- Swartzwelter, B.; Fux, A.; Johnson, L.; Swart, E.; Hofer, S.; Hofstätter, N.; Geppert, M.; Italiani, P.; Boraschi, D.; Duschl, A.; et al. The Impact of Nanoparticles on Innate Immune Activation by Live Bacteria. Int. J. Mol. Sci. 2020, 21, 9695.
- Herrera, M.G.; Nicoletti, F.; Gras, M.; Dörfler, P.W.; Tonali, N.; Hannappel, Y.; Ennen, I.; Hütten, A.; Hellweg, T.; Lammers, K.M.; et al. Pepsin Digest of Gliadin Forms Spontaneously Amyloid-Like Nanostructures Influencing the Expression of Selected Pro-Inflammatory, Chemoattractant, and Apoptotic Genes in Caco-2 Cells: Implications for Gluten-Related Disorders. Mol. Nutr. Food Res. 2021, 65, 2100200.
- Maiuri, L.; Ciacci, C.; Ricciardelli, I.; Vacca, L.; Raia, V.; Auricchio, S.; Picard, J.; Osman, M.; Quaratino, S.; Londei, M. Association between innate response to gliadin and activation of pathogenic T cells in coeliac disease. Lancet 2003, 362, 30–37.
- Londei, M.; Ciacci, C.; Ricciardelli, I.; Vacca, L.; Quaratino, S.; Maiuri, L. Gliadin as a stimulator of innate responses in celiac disease. Mol. Immunol. 2005, 42, 913–918.
- Qiao, S.-W.; Bergseng, E.; Molberg, Ø.; Xia, J.; Fleckenstein, B.; Khosla, C.; Sollid, L.M. Antigen Presentation to Celiac Lesion-Derived T Cells of a 33-Mer Gliadin Peptide Naturally Formed by Gastrointestinal Digestion. J. Immunol. 2004, 173, 1757–1762.
- Fraser, J.S.; Engel, W.; Ellis, H.J.; Moodie, S.J.; Pollock, E.L.; Wieser, H.; Ciclitira, P.J. Coeliac disease: In vivo toxicity of the putative immunodominant epitope. Gut 2003, 52, 1698–1702.
- Sollid, L.M. Intraepithelial Lymphocytes in Celiac Disease: License to Kill Revealed. Immunity 2004, 21, 303–304.
- Herrera, M.G.; Zamarreño, F.; Costabel, M.; Ritacco, H.; Hütten, A.; Sewald, N.; Dodero, V.I. Circular dichroism and electron microscopy studies in vitro of 33-mer gliadin peptide revealed secondary structure transition and supramolecular organization. Biopolymers 2014, 101, 96–106.
- Herrera, M.G.; Benedini, L.; Lonez, C.; Schilardi, P.L.; Hellweg, T.; Ruysschaert, J.-M.; Dodero, V.I. Self-assembly of 33-mer gliadin peptide oligomers. Soft Matter 2015, 11, 8648–8660.
- Herrera, M.; Vazquez, D.; Sreij, R.; Drechsler, M.; Hertle, Y.; Hellweg, T.; Dodero, V. Insights into gliadin supramolecular organization at digestive pH 3.0. Colloids Surf. B Biointerfaces 2018, 165, 363–370.
- Herrera, M.G.; Veuthey, T.V.; Dodero, V.I. Self-organization of gliadin in aqueous media under physiological digestive pHs. Colloids Surf. B Biointerfaces 2016, 141, 565–575.
- Herrera, M.G.; Pizzuto, M.; Lonez, C.; Rott, K.; Hütten, A.; Sewald, N.; Ruysschaert, J.-M.; Dodero, V.I. Large supramolecular structures of 33-mer gliadin peptide activate toll-like receptors in macrophages. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 1417–1427.
- Amundarain, M.J.; Herrera, M.G.; Zamarreño, F.; Viso, J.F.; Costabel, M.D.; Dodero, V.I. Molecular mechanisms of 33-mer gliadin peptide oligomerisation. Phys. Chem. Chem. Phys. 2019, 21, 22539–22552.
- Falcigno, L.; Calvanese, L.; Conte, M.; Nanayakkara, M.; Barone, M.V.; D’Auria, G. Structural Perspective of Gliadin Peptides Active in Celiac Disease. Int. J. Mol. Sci. 2020, 21, 9301.
- Nanayakkara, M.; Lania, G.; Maglio, M.; Auricchio, R.; De Musis, C.; Discepolo, V.; Miele, E.; Jabri, B.; Troncone, R.; Auricchio, S.; et al. P31–43, an undigested gliadin peptide, mimics and enhances the innate immune response to viruses and interferes with endocytic trafficking: A role in celiac disease. Sci. Rep. 2018, 8, 1–12.
- Herrera, M.G.; Castro, M.F.G.; Prieto, E.; Barrera, E.; Dodero, V.I.; Pantano, S.; Chirdo, F. Structural conformation and self-assembly process of p31-43 gliadin peptide in aqueous solution. Implications for celiac disease. FEBS J. 2019, 287, 2134–2149.
- Castro, M.F.G.; Miculán, E.; Herrera, M.G.; Ruera, C.; Perez, F.; Prieto, E.D.; Barrera, E.; Pantano, S.; Carasi, P.; Chirdo, F.G. p31-43 Gliadin Peptide Forms Oligomers and Induces NLRP3 Inflammasome/Caspase 1- Dependent Mucosal Damage in Small Intestine. Front. Immunol. 2019, 10, 31.
- Oldstone, M.B.A. Molecular mimicry and immune-mediated diseases. FASEB J. 1998, 12, 1255–1265.
- Kohm, A.P.; Fuller, K.G.; Miller, S.D. Mimicking the way to autoimmunity: An evolving theory of sequence and structural homology. Trends Microbiol. 2003, 11, 101–105.
- Rojas, M.; Restrepo, P.; Monsalve, D.M.; Pacheco, Y.; Acosta-Ampudia, Y.; Ramírez-Santana, C.; Leung, P.S.; Ansari, A.A.; Gershwin, M.E.; Anaya, J.-M. Molecular mimicry and autoimmunity. J. Autoimmun. 2018, 95, 100–123.
- Gómez-Rial, J.; Calle, I.R.; Salas, A.; Martinón-Torres, F. Rotavirus and autoimmunity. J. Infect. 2020, 81, 183–189.
- Cuan-Baltazar, Y.; Soto-Vega, E. Microorganisms associated to thyroid autoimmunity. Autoimmun. Rev. 2020, 19, 102614.
- Múnera, M.; Farak, J.; Pérez, M.; Rojas, J.; Villero, J.; Sánchez, A.; Emiliani, Y. Prediction of molecular mimicry between antigens from Leishmania sp. and human: Implications for autoimmune response in systemic lupus erythematosus. Microb. Pathog. 2020, 148, 104444.
- Cunningham, M.W. Streptococcus and rheumatic fever. Curr. Opin. Rheumatol. 2012, 24, 408–416.
- Cunningham, M.W. Molecular Mimicry, Autoimmunity, and Infection: The Cross-Reactive Antigens of Group A Streptococci and their Sequelae. Microbiol. Spectr. 2019, 7.
- Lucchese, G.; Flöel, A. SARS-CoV-2 and Guillain-Barré syndrome: Molecular mimicry with human heat shock proteins as potential pathogenic mechanism. Cell Stress Chaperones 2020, 25, 731–735.
- Ozuna, C.V.; Iehisa, J.C.M.; Gimenez, M.J.; Alvarez, J.B.; Sousa, C.; Barro, F. Diversification of the celiac disease α-gliadin complex in wheat: A 33-mer peptide with six overlapping epitopes, evolved following polyploidization. Plant J. 2015, 82, 794–805.
- Kurochkina, N.; Guha, U. SH3 domains: Modules of protein–protein interactions. Biophys. Rev. 2013, 5, 29–39.
- Teyra, J.; Huang, H.; Jain, S.; Guan, X.; Dong, A.; Liu, Y.; Tempel, W.; Min, J.; Tong, Y.; Kim, P.M.; et al. Comprehensive Analysis of the Human SH3 Domain Family Reveals a Wide Variety of Non-canonical Specificities. Struct. 2017, 25, 1598–1610.e3.
- Chen, H.I.; Sudol, M. The WW domain of Yes-associated protein binds a proline-rich ligand that differs from the consensus established for Src homology 3-binding modules. Proc. Natl. Acad. Sci. USA 1995, 92, 7819–7823.
- Ball, L.J.; Kühne, R.; Schneider-Mergener, J.; Oschkinat, H. Recognition of Proline-Rich Motifs by Protein-Protein-Interaction Domains. Angew. Chem. Int. Ed. 2005, 44, 2852–2869.
- Bork, P.; Sudol, M. The WW domain: A signalling site in dystrophin? Trends Biochem. Sci. 1994, 19, 531–533.
- Omasits, U.; Ahrens, C.; Müller, S.; Wollscheid, B. Protter: Interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics 2014, 30, 884–886.
- Méthot, P.-O.; Alizon, S. What is a pathogen? Toward a process view of host-parasite interactions. Virulence 2014, 5, 775–785.
- Ingram, J.H.; Stone, M.; Fisher, J.; Ingham, E. The influence of molecular weight, crosslinking and counterface roughness on TNF-alpha production by macrophages in response to ultra high molecular weight polyethylene particles. Biomaterials 2004, 25, 3511–3522.
- Matthews, J.; Green, T.R.; Stone, M.H.; Wroblewski, B.M.; Fisher, J.; Ingham, E. Comparison of the response of primary human peripheral blood mononuclear phagocytes from different donors to challenge with model polyethylene particles of known size and dose. Biomaterials 2000, 21, 2033–2044.
- Green, T. Polyethylene particles of a ‘critical size’ are necessary for the induction of cytokines by macrophages in vitro. Biomaterials 1998, 19, 2297–2302.
- Paul, D.; Achouri, S.; Yoon, Y.-Z.; Herre, J.; Bryant, C.E.; Cicuta, P. Phagocytosis Dynamics Depends on Target Shape. Biophys. J. 2013, 105, 1143–1150.
- Bascuñán, K.A.; Araya, M.; Roncoroni, L.; Doneda, L.; Elli, L. Dietary Gluten as a Conditioning Factor of the Gut Microbiota in Celiac Disease. Adv. Nutr. 2019, 11, 160–174.
- Akobeng, A.K.; Singh, P.; Kumar, M.; Al Khodor, S. Role of the gut microbiota in the pathogenesis of coeliac disease and potential therapeutic implications. Eur. J. Nutr. 2020, 59, 3369–3390.
- Polo, A.; Arora, K.; Ameur, H.; Di Cagno, R.; De Angelis, M.; Gobbetti, M. Gluten-free diet and gut microbiome. J. Cereal Sci. 2020, 95, 103058.
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Health Dis. 2015, 26, 26191.