1. Introduction
The idea of using immune response against abnormal cells in the body to treat cancer has been tested in the past few decades and evolved from using recombinant cytokines to adoptive cell transfer [1][2][1,2]. The first generation of immunotherapies like high-dose interleukin-2 were limited by low response rates and high incidence of serious adverse events, but the durability of response encouraged further research in the field [3][4][5][3,4,5]. Discovery of checkpoints of T-cell activation and development of monoclonal antibodies targeting the checkpoints dramatically changed the outcomes of immunotherapy [6][7][8][9][10][11][12][6,7,8,9,10,11,12]. Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1) were the early targets that were discovered and characterized in the late 1980s and early 1990s, respectively [13][14][15][16][17][18][19][13,14,15,16,17,18,19]. Both CTLA-4 and PD-1 have been shown to be reliable targets, and to date, seven drugs have been approved for different types of cancers, such as melanoma and lung cancer [20][21][22][23][24][20,21,22,23,24]. In addition to monotherapy, combination of CTLA-4 and PD-1 blockers is also approved for treatment of multiple cancer types [23]. While the CTLA-4 and PD-1 blockers had decent and durable response rates, a large fraction of patients did not respond to the treatment, and the incidence of serious adverse events was high in the responding patients [25][26][27][25,26,27]. The need for safer targets that can be blocked or activated to achieve reasonable anti-tumor response with manageable adverse events and that can be combined with PD-1/PD-L1 blockers or other immune checkpoint blockers led to the identification of T-cell immunoglobulin and ITIM domain (TIGIT), an inhibitory immune checkpoint, and the development of anti-TIGIT antibodies.
TIGIT is considered as an important target mainly because of its expression profile (natural killer cells (NK cells), cytotoxic CD8 + T cells and regulatory T cells (Tregs) [28]. More importantly, the phenotype of Tigit −/− mouse was reported to be mild, and the knockout mice did not spontaneously develop autoimmunity, indicating a comparatively milder safety profile [29].
2. Tigit
TIGIT expression is mainly seen on resting CD4 + CD25 hi Treg cells, activated T cells, NK cells, NKT cells, and memory T cells. Naïve CD4 + T cells do not express TIGIT, but its expression is induced at mRNA levels upon activation [30][35]. TIGIT has been reported as marker for CD8+ T-cell exhaustion and is also a characteristic marker for Tregs in the tumor microenvironment [7][28][29][31][32][7,28,29,37,38].
CD155 expression is mainly reported on DCs, T cells, B cells, and macrophages, whereas CD112 is widely expressed on both hematopoietic and non-haematopoietic tissues, including bone marrow, lung, pancreas, and kidney [33][34][40,41]. CD113 expression is limited to non-hematopoietic tissues, such as lung, liver, testis, kidney, and placenta [35][42]. Several human cancers are reported to overexpress CD155 and CD112 [36][37][38][43,44,45]. Interestingly, interferon-γ (IFN- γ) was shown to up-regulate the expression of CD155 on human vascular endothelial cells, a mechanism similar to induction of PD-1/PD-L1 pathway [39][46].
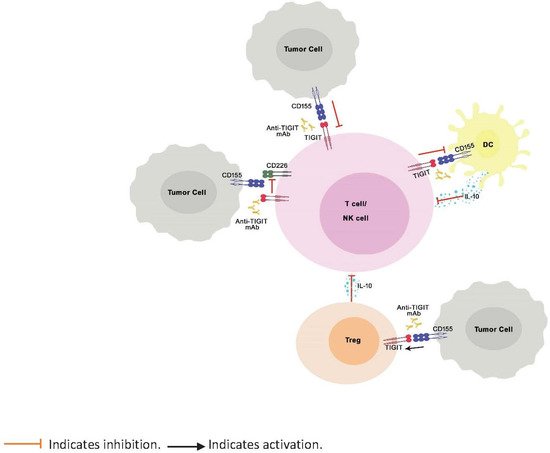
TIGIT is a negative regulator of immune response known to bind to PVR ligands with greater affinity and outcompete the costimulatory receptors, CD226 and CD96, expressed on T cells, thereby inhibiting the activation, proliferation, and differentiation of T cells ( Figure 1 ). Further, TIGIT engagement ensures the survival of inhibited T cells by activating cell survival pathways. [7]. TIGIT activation on NK cells was shown to inhibit cytotoxic granule polarization and IFN-γ production and decrease NK cell cytotoxicity [40][41][30,47]. In addition, TIGIT interaction on Tregs skews the cytokine balance, suppresses Th1 or Th17 phenotype, and induces Th2 phenotype [29][42][29,48]. However, unlike CTLA-4 and PD-1, which, when knocked out in mice, are known to manifest as severe and spontaneous autoimmune phenotype [17][43][44][45][17,49,50,51], TIGIT knock-out mice do not spontaneously develop autoimmune phenotype, indicating mild to moderate control of TIGIT over immune response [29].
Figure 1. Role of TIGIT in regulation of immune response. TIGIT competitively inhibits binding of CD226 to CD155 and impairs the CD226-mediated activation of T cells and NK cells. Binding of TIGIT to its ligand CD155 results in activation of inhibitory signals in T cells and NK cells. TIGIT binding to CD155 on APCs results in IL-10 production, decreased IL-12 production (not shown in the illustration), and indirect inhibition of T cells. Finally, TIGIT signaling enhances the immunosuppressive functions of Tregs.
Even before the discovery of TIGIT, its ligands were known to be upregulated on the surface of tumor cell surface. Expression of nectin family of proteins and their role in cell adhesion and survival was reported in tumors from epithelial origin, such as non-small cell lung cancer, colon cancer, and metastatic neuroblastoma, and also tumors from hematopoietic origin, such as myeloid leukemia [36][46][47][48][49][43,52,53,54,55]. High expression of CD155 was shown to be an independent prognostic marker and predictor of poor clinical outcome in breast cancer patients [50][56]. The recently discovered ligand for TIGIT, nectin 4, was shown to be overexpressed in breast, bladder, lung, and pancreatic cancers [51][57]. On the other hand, TIGIT expression was also reported to be upregulated on lymphocytes in tumor microenvironment. Studies showed TIGIT expression on CD8 + , CD4 + T cells, and NK cells paralleled to that of PD-1 in hepatocellular, lung, and colorectal cancers and in Hodgkin’s lymphoma [52][53][54][55][56][57][58][59][58,59,60,61,62,63,64,65].
3. Anti-Tigit Antibodies in Development
Targeting TIGIT-PVR pathway has gained importance in the recent months, and several biotech/pharmaceutical companies are working on development of anti-TIGIT antibodies. As of June 2020, 15 antibodies targeting TIGIT-PVR pathway are being commercially developed and are in various stages of clinical development. The list of molecules, details of antibody isotypes, Fc status, and current status is presented in
Table 13. Nine molecules are in clinical trials, and tiragolumab, developed by Genentech and ociperlimab, developed by BeiGene, are in the most advanced stage of development (Phase III). In January 2021, FDA granted breakthrough therapy designation, a pathway designed to accelerate the development and review of data for tiragolumab plus atezolizumab combination as first-line treatment of people with metastatic non-small cell lung cancer (NSCLC) whose tumors have high PD-L1 expression with no EGFR or ALK genomic tumor aberrations
[60][70]. Nearly half of the antibodies developed have IgG1 back bone, whereas the antibody developed by Astellas Pharma has IgG4 back bone (
Table 13).
Table 13.
List of anti-TIGIT molecules in clinical development.
| Generic Name |
Type |
FcγR Status |
Company |
Status |
| Tiragolumab (MTIG7192A) |
Fully human IgG1 |
Active |
Genentech |
Phase III |
| Ociperlimab (BGB-A1217) |
Humanized IgG1 |
Active |
BeiGene USA, Inc |
Phase III |
| Vibostolimab (MK-7684) |
Fully human IgG1 |
Active |
Merck & Co Inc |
Phase II |
| Domvanalimab (AB-154) |
Fully human IgG1 |
Inactive |
Arcus Biosciences Inc |
Phase II |
Several factors, such as origin of the antibody backbone (mouse, chimeric, humanized, or fully human), IgG backbone for the antibody, FcγR binding status, and dose, play a key role in the development and eventual clinical success of the antibody. Details of the factors are discussed in the following sections.
3.2. Origin
The origin of antibodies intended for therapeutic application can significantly impact the clinical success of the molecules. Antibodies generated in mice were shown to induce production of anti-mouse antibodies in patients, which increased the clearance of antibody-based drugs. Chimeric antibodies, which have part of their protein structure from human origin and the other part from animal origin, were expected to be better but still suffered due to anti-drug antibodies. Humanized antibodies, which have protein sequences that closely matched to that of humans and fully human antibodies that do not have any protein sequence from mouse (or other animal) origin, are expected to be least immunogenic and have very low chances of anti-drug antibody development
[62][63][72,73]. All the approved immune checkpoint blockers to date are either humanized (pembrolizumab, atezolizumab) or fully human (ipilimumab, nivolumab), and the majority of the anti-TIGIT antibodies in clinical development are fully human antibodies (
Table 13).
3.3. IgG Isotype and FcγR Binding
Almost all the commercially developed immune checkpoint blocking antibodies have IgG backbone. IgG based antibodies are known to interact with FcγR on innate effector immune cells through their Fc-region and induce antibody dependent cellular cytotoxicity (ADCC) in the target cells. ADCC is a non-phagocytic mechanism through which innate immune cells including macrophages, DCs, neutrophils, and NK cells, kill the antibody-bound target cells
[7][64][65][66][67][68][7,74,75,76,77,78]. Activation of ADCC through the binding of Fc-γRI (CD64), Fc-γ RIIa, Fc-γ RIIc (CD32), and Fc-γ RIIIa (CD16) triggers the release of cytotoxic mediators, such as tumor necrosis factor-α (TNF-α), perforin, granzyme, and reactive oxygen species (ROS), from effector immune cells on to the target cell surface and result in lysis of target cells.
Affinity of the antibody to Fcγ receptors of effector cells is the key to induction of ADCC and is mainly dependent on the antibody backbone. IgG1 backbone has highest affinity to all the three stimulatory FcγRs and induces significant ADCC, whereas IgG2 backbone does not bind to FcγRs and does not induce ADCC
[7]. ADCC is the most common factor that is considered during the development of therapeutic antibodies. Induction of ADCC is a desired effect for antibodies targeting receptors on cancer cells and has been shown to be a contributor to the anti-tumor activity of monoclonal antibodies
[69][70][71][79,80,81].
While ADCC is considered beneficial for the antibody-drug conjugates, its contribution to the activity of immune checkpoint blockers is not completely clear. For example, PD-1-blocking antibodies pembrolizumab and nivolumab that showed promising success in the treatment of cancer have IgG4 backbone and are known to have relatively lower binding affinity to FcγRs. They also did not show significant ADCC activity in the in-vitro models
[72][82]. Similarly, tislelizumab, an anti-PD-1 antibody with IgG4 backbone is specifically designed to minimize FcγRs
[73][74][75][83,84,85]. However, the anti-CTLA-4 antibody, ipilimumab, which is IgG1 based and known to induce ADCC, has been successful compared to IgG2 based anti-CTLA-4 antibody, tremelimumab, which does not have ADCC activity.
The FcγR binding region of anti-TIGIT antibodies in clinical development is active in some of the molecules and inactivated in others. Based on publicly available information, six out of nine molecules, including tiragolumab, ociperlimab, vibostolimab, EOS-448, etigilimab, and AGEN-1307, have active FcγR binding region, whereas three molecules, including domvanalimab, BMS-986207, and CASC-674, have inactive FcγR binding region (
Table 13). The FcγR binding region in AGEN-1307 is mutated to enhance the binding of the antibody with Fcγ receptors and increase its ADCC activation
[76][86]. It remains to be seen if the presence or absence of FcγR binding region in the antibody would have an impact on the clinical efficacy of anti-TIGIT antibodies.
3.4. Dose
Various factors, including target binding affinity; pharmacodynamic factors, like saturation of downstream biomarker response and concentration at which optimal receptor occupancy is achieved; pharmacokinetic factors, like saturation of target-mediated elimination pathway and anti-drug antibodies that reduce target drug concentration, dose, or exposure-response relationships for efficacy and safety; and maximum tolerated dose and dose at which drug is expected to have maximum effect, are considered before selecting the dose for advanced studies. PK-PD models, simple mechanistic models, as well as complex mechanistic models, such as quantitative systems pharmacology models, are typically used to simulate the dose that achieves optimal target occupancy and achieves desired pharmacological effect. In cases where information is not completely available to develop PK-PD models or mechanistic models, available literature information from related molecules is used to propose the dose that can possibly have desired response.
Results from Phase I studies across multiple clinical programs demonstrate that anti-TIGIT antibodies are well tolerated (
Table 24). Studies used different dose ranges of the antibody and used either every two weeks (Q2W) or every three weeks (Q3W) administration regimen. Dose-limiting toxicities were not recorded during monotherapy or in combination with anti-PD-1 antibody for any of the anti-TIGIT antibodies in clinical development, indicating molecules against this target have broad therapeutic index. Highest dose of anti-TIGIT antibody evaluated was 20 mg/kg Q2W for etigilimab (
Table 24). Clinical activity (objective response rate) observed after anti-TIGIT antibody monotherapy was minimal to none, indicating combination therapy with anti-PD1 or PD-L1 or other agents is needed. Complete peripheral TIGIT receptor occupancy was observed for most drugs at very low doses. For example, tiragolumab evaluated doses starting at 2 mg, and complete receptor occupancy was observed at 30 mg dose. Similarly, ociperlimab evaluated doses starting at 50 mg, and complete receptor occupancy was observed at this dose and above. Domvanalimab reported complete receptor occupancy at the dose of 0.5 mg/kg
[77][87].
Table 24.
Studies reporting anti-TIGIT antibody dose and tolerability.
| Drug |
Phase |
Dose and Regimen |
Comment |
Reference |
| Tiragolumab |
Phase III
Multiple Solid tumors |
2 mg to 1200 mg Q3W
RP2D: 600 mg Q3W |
100% receptor occupancy seen at ≥30 mg and clinical activity observed at doses 400 mg to 600 mg.
600 mg Q3W was proposed as dose for Ph2 study. |
Bendell et al. AACR 2020 |
| Ociperlimab |
Phase III |
50 mg to 900 mg
RP2D: 900 mg Q3W |
100% receptor occupancy was observed at 50 mg, and linear PK was observed through 900 mg. |
NCT04746924 |
| Domvanalimab (AB-154) |
Phase I
NSCLC |
0.5 mg/kg; 1 mg/kg & 3 mg/kg Q2W |
100% receptor occupancy seen at 3 mg/kg |
Anderson et al. SITC 2019 p260 |
| Vibostolimab (MK-7684) |
Phase I
Multiple Solid tumors |
2.1 mg to 700 mg Q3W
RP2D: 200 mg Q3W |
ORR 19% in combination with pembrolizumab
Vibostolimab well tolerated as monotherapy and in combination with 200 mg pembrolizumab |
Golan et al. SITC 2018 |
| BMS-986207 |
Fully human IgG1 |
Inactive |
Bristol-Myers Squibb Co |
Phase II |
| BMS-986207 |
Phase I/II |
Not disclosed |
No details |
NCT02913313 |
EOS-448 |
| EOS-448 | Fully human IgG1 |
Active |
iTeos Therapeutics SA |
Phase II |
| Phase I |
0.1 mg/kg, 1 mg/kg and 10 mg/kg |
Receptor occupancy increased with dose. Nearly 100% occupancy was seen at 10 mg/kg dose. Dose-limiting toxicity was not seen. |
Nguyen et al. AACR 2020 |
ASP-8374 |
Fully human IgG4 |
Inactive |
Astellas Pharma Inc |
Phase I |
| ASP-8374 |
Phase I
Solid tumors |
Not disclosed |
Details not available |
NCT03260322 |
COM-902 |
Mouse/cyno cross-reactive fully human IgG1 antibody |
NA |
Compugen Ltd. |
| COM-902 |
Phase I
Solid tumors | Phase I |
| 7 doses to be tested for dose limiting toxicity. |
| Q3W regimen |
Data not available. Study posted in April 2020. |
NCT04354246 |
Etigilimab |
Fully human IgG1 |
Active |
Mereo Biopharma Group Plc |
Phase I |
| Etigilimab |
Phase I |
0.3 mg/kg to 20 mg/kg Q2W |
Safely administered up to 20 mg/kg. Stable disease was seen in 7/18 patients across all doses |
IBI-939 |
NA |
NA |
Innovent Biologics Inc |
IND Filed |
| Sharma et al. SITC 2018 |
AGEN-1307 |
Fully human IgG1 |
Active and enhanced |
Agenus Inc |
Preclinical |
| CASC-674 |
Fully human IgG2a |
Inactive |
Seattle Genetics Inc |
Preclinical |
| Anti-PVR Antibody (NB-6253) |
NA |
NA |
Northern Biologics Inc |
Preclinical |
| PH-804 |
NA |
NA |
Phio Pharmaceuticals Corp |
Preclinical |
| TIGIT-PD-L1 dual |
NA |
NA |
Aurigene Discovery Technologies Ltd. |
Preclinical |
NA, details not available.
In addition to monospecific antibodies, researchers are also developing bispecific antibody that co-targets PD-L1 and TIGIT. Generation and characterization of a multivalent bispecific antibody consisting of tetravalent anti-PD-L1 Fc-fusion nanobody and tetravalent anti-TIGIT nanobody was recently reported
[61][71]. The bispecific antibody showed high specificity and affinity to primate PD-L1 and TIGIT and had significantly higher anti-tumor activity compared to PD-L1 antibody in mouse models. Benefits of bispecific antibodies targeting multiple immune checkpoints need to be demonstrated in clinical studies.
3.1. Factors Considered during Development
Based on the concentration at which maximum receptor occupancy was achieved, and based on the concentration at which early clinical activity was noticed, a dose of 600 mg was proposed for tiragolumab phase II studies
[78][88]. Tiragolumab’s recommended phase II dose (RP2D) of 600 mg Q3W is approximately 20-fold higher than the initial dose at which complete peripheral receptor occupancy was observed. Similarly, ociperlimab RP2D was 900 mg Q3W in published clinical trials, which is ~18-fold higher than the initial dose at which complete receptor occupancy was observed (NCT04746924). Vibostolimab appears to be investigating RP2D of 200 mg Q3W based on the clinical study posted (NCT04738487); however, no information-receptor on occupancy or other pharmacodynamic biomarker data are available. Though other molecules, including domvanalimab, BMS986207, and EOS-448, also entered into phase II trials (
Table 13), dose of the antibody has not been publicly disclosed at the time of data compilation.
3.5. Safety
Anti-TIGIT antibodies were found to be generally well tolerated when administered as monotherapy as well as when administered in combination with PD-1/PD-L1 blockers (Table 24). Most common adverse events reported in more than 10% patients included fatigue and pruritus; both were Grade 1. Two Grade 2 events, anemia and diarrhea, were reported in two patients treated with vibostolimab monotherapy. There were no Grade 3–5 events reported with anti-TIGIT antibody monotherapy.