Atopic dermatitis (AD) is an eczematous, pruritic skin disorder with extensive barrier dysfunction and elevated interleukin (IL)-4 and IL-13 signatures. The barrier dysfunction correlates with the downregulation of barrier-related molecules such as filaggrin (FLG), loricrin (LOR), and involucrin (IVL). IL-4 and IL-13 potently inhibit the expression of these molecules by activating signal transducer and activator of transcription (STAT)6 and STAT3. In addition to IL-4 and IL-13, IL-22 and IL-17A are probably involved in the barrier dysfunction by inhibiting the expression of these barrier-related molecules. In contrast, natural or medicinal ligands for aryl hydrocarbon receptor (AHR) are potent upregulators of FLG, LOR, and IVL expression. As IL-4, IL-13, IL-22, and IL-17A are all capable of inducing oxidative stress, antioxidative AHR agonists such as coal tar, glyteer, and tapinarof exert particular therapeutic efficacy for AD. These antioxidative AHR ligands are known to activate an antioxidative transcription factor, nuclear factor E2-related factor 2 (NRF2). This article focuses on the mechanisms by which FLG, LOR, and IVL expression is regulated by IL-4, IL-13, IL-22, and IL-17A. The author also summarizes how AHR and NRF2 dual activators exert their beneficial effects in the treatment of AD.
- atopic dermatitis
- skin barrier
- filaggrin
- filaggrin-2
- loricrin
- involucrin
- IL-4
- IL-13
- IL-17A
- IL-22
Atopic dermatitis (AD) is an eczematous, pruritic skin disorder with extensive barrier dysfunction and elevated interleukin (IL)-4 and IL-13 signatures. The barrier dysfunction correlates with the downregulation of barrier-related molecules such as filaggrin (FLG), loricrin (LOR), and involucrin (IVL). IL-4 and IL-13 potently inhibit the expression of these molecules by activating signal transducer and activator of transcription (STAT)6 and STAT3. In addition to IL-4 and IL-13, IL-22 and IL-17A are probably involved in the barrier dysfunction by inhibiting the expression of these barrier-related molecules. In contrast, natural or medicinal ligands for aryl hydrocarbon receptor (AHR) are potent upregulators of FLG, LOR, and IVL expression. As IL-4, IL-13, IL-22, and IL-17A are all capable of inducing oxidative stress, antioxidative AHR agonists such as coal tar, glyteer, and tapinarof exert particular therapeutic efficacy for AD. These antioxidative AHR ligands are known to activate an antioxidative transcription factor, nuclear factor E2-related factor 2 (NRF2).
1. Introduction
- Introduction
The human epidermis is composed of stratified layers, differentiating from the basal layer on the basement membrane, towards spinous and granular layers, and finally to the outermost cornified layer [1][2][3][4][5][2][1–6]. Keratinocytes are the major constituent of epidermal cells. In this stratification process, the keratinocytes undergo well-orchestrated differentiation to optimize the skin barrier to survive the dry, harsh terrestrial conditions. Basal layer keratinocytes, expressing keratin 5 (K5) and K14, divide and move up to the spinous layer. Spinous layer keratinocytes then switch their keratin profile to K1 and K10. Keratins connect to the desmosomes, which are the cell–cell adhesion structures specific for stratified epithelium [1][2][3][4][5][2][1–6]. In the granular layer, keratinocytes commit to synthesize keratohyalin granules, which are primarily composed of profilaggrin and loricrin (LOR) [1][2][3][4][5][2][1–6]. This process coincides with the decreases of K1 and K10, increase of calcium and activation of protein kinase C [6][2]. In normal orthokeratotic conditions, cells are denucleated when the uppermost keratinocytes in the granular layer turn into cornified cells (corneocytes) in the cornified layer. Profilaggrin is processed to filaggrin (FLG) repeats and the cleaved N-terminus of profilaggrin translocates into the nucleus and may trigger the denucleation process [7][8][9][7–9]. The cornified cells provide a specialized cell membrane called the cornified envelope. The cornified envelope is composed of various cytoskeletal and barrier-related molecules, including K1, K10, desmosomal proteins (envoplakin and periplakin), LOR, FLG, filaggrin-2 (FLG2), and involucrin (IVL), which are crosslinked by transglutaminase 1 and partly by transglutaminases 3 and 5 [1][2][3][4][5][2][1–6]. Granular layer keratinocytes also produce lamellar granules, which are rich in polar lipids, glycosphingolipids, free sterols, and phospholipids. The lamellar granule lipids are released and integrated into the intercellular space lipids containing sterols, fatty acids and ceramides in the cornified layer. Some ceramides with ultralong omega-hydroxyl chains are covalently bound to the outside of the cornified envelope via scaffolds of IVL, envoplakin and periplakin [1][2][3][4][5][2][1–6]. This thin lipid layer covering the cornified envelope is called the corneocyte-bound lipid envelope [2][10][6,10]. These three structures, cornified envelope, corneocyte-bound lipid envelope, and intercellular space lipids, are key players in maintaining appropriate barrier function [1][2][3][4][5][2][1–6].
Upon terminal differentiation, keratinocytes synthesize a number of barrier-related molecules, which are sequentially incorporated into the above-mentioned three cardinal barrier structures. Notably, many barrier-related molecules expressed in the granular layer are genetically mapped to the chromosome 1q21.3 locus, which is called the epidermal differentiation complex (EDC) [4][11][5,11]. FLG, FLG2, LOR, and IVL genes are all located in the EDC [11][5,11]. The EDC also includes a series of genes encoding S100A proteins, such as S100A7 [12][13][5,12,13]. Most S100A proteins exert antimicrobial and proinflammatory effects [12][5,12]. LOR, IVL, late cornified envelope protein genes (LCEs), and small proline-rich protein genes (SPRRs) are classified into the “cornified envelope precursor gene family” [6][5]. FLG, FLG2, and hornerin (HRNR) genes are classified into the “fused gene family” evolved from the “S100A protein gene family” and the “cornified envelope precursor gene family” [6][5] (Figure 1).
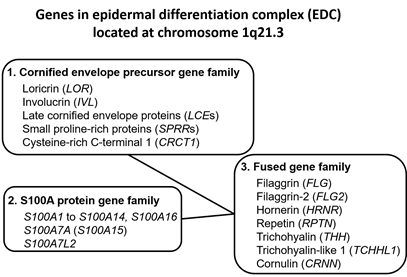
Figure 1. Genes encoding the epidermal differentiation complex.
The expression of EDC genes is not stable, but is actively modulated by external stimuli, including ultraviolet irradiation and photoproducts [14][15][14,15], dioxins, and other oxidative pollutants [16][4,16], bioproducts of commensal or symbiotic microorganisms such as Malassezia and Staphylococcus epidermidis [17][18][19][17–19], cosmetics [20], and various phytochemicals [21][22][23][24][25][21–25]. These chemical stimulants activate the xenobiotic chemical sensor aryl hydrocarbon receptor (AHR), upregulate the expression of barrier-related proteins, and accelerate the terminal differentiation of keratinocytes [26][4,14,16,18,26]. Consistent with this, long-lasting activation of AHR by dioxins induces the exaggerated terminal differentiation of keratinocytes and sebocytes, leading to the development of chloracne [27][28][29][27–29]. In particular, the exaggerated AHR activation converts sebocyte differentiation from sebaceous cell differentiation to keratinocytic differentiation, which results in the loss of sebocytes and keratinous cyst formation [29][30][29,30]. This is probably the major cause of chloracne. Although the mechanisms of accelerated keratinization are not fully understood, intracellular levels of reactive oxygen species (ROS) are one of the major modulators [31][32][33][34][35][36][37][38][4,31–38]. The intracellular oxidative stress level is fine-tuned by the oxidative stress-prone AHR system [31][39][4,31,39] and the antioxidative nuclear factor E2-related factor 2 (NRF2) system [32][33][34][35][36][37][38][40][41][42][43][32–38,40–43].
The expression of EDC genes is also actively modulated by internal stimuli such as cytokines. The gene expression of FLG, FLG2, LOR, IVL, and S100A7 is differentially affected in atopic dermatitis (AD) and psoriasis [44][45][44,45]. The dysregulated expression of FLG, FLG2, LOR, and IVL is known to be normalized by specific biologic treatments, for example, blockade of interleukin-4 (IL-4) and IL-13 in AD [44] or blockade of IL-17A in psoriasis [45]. The levels of IL-22 are elevated in both AD [46][47][46,47] and psoriasis [48][49][48,49]. IL-22 is also known to modulate the gene expression of these barrier-related molecules [50][51][52][50–52]. As the expression of IVL, LOR, FLG, FLG2, and other EDC genes is differentially altered by these pathogenic cytokines, the expression of these molecules is commonly used as reliable markers for evaluating the therapeutic efficacy of relevant biologics [44][45][44,45].
2. Roles of IVL, LOR, FLG, and FLG2 in Epidermal Barrier Formation
- Roles of IVL, LOR, FLG, and FLG2 in Epidermal Barrier Formation
IVL has high structural homology with LOR in the glutamine- and lysine-rich amino- and carboxy-terminal domains [5]. IVL is expressed in the upper spinous layer, but mainly in the granular layers, and is involved in the initial step of cornified envelope formation. Cornified envelope formation starts from desmosomes where IVL is crosslinked with envoplakin, periplakin, and keratin filaments by transglutaminase 1 [5,6]. This protein complex also becomes the scaffold for the corneocyte-bound lipid envelope [6,10].
LOR is the most abundant component of the cornified envelope [1,3,5]. It is very hydrophobic, insoluble, and is easily polymerized via disulfide crosslinking in ambient air, making it suitable as a protein that reinforces the cornified envelope [1,5]. LOR is expressed in the granular layer and is crosslinked to IVL, envoplakin, and periplakin scaffolds by transglutaminase 1 [1,5].
Profilaggrin consists of a conserved small N-terminal domain, 10–12 FLG repeats and a C-terminal domain [5]. Profilaggrin to FLG processing requires several proteases, such as profilaggrin endopeptidase 1, matriptase 1, and channel-activating protease 1. FLG is involved in aggregating the K1 and K10 filaments into higher-molecular-weight parallel structures that facilitate the incorporation of K1 and K10 into the cornified envelope and contribute to the thin granular keratinocyte shape [53][54][55][56][1,5,53]. FLG peptides are simultaneously degraded by caspase 14 and calpain 1 into free hydrophilic amino acids, which maintain the intracellular water content [1,5,6]. Ichthyosis vulgaris is caused by the loss-of-function mutation of FLG [57][54]. Loss-of-function mutations of FLG have been demonstrated in a subpopulation of patients with AD, at rates ranging from 10% to 50% depending on the ethnicity [58][59][55,56]. Therefore, AD is a significant comorbidity with ichthyosis vulgaris [60][61][57,58].
FLG2 contains two distinct repeat domains, A and B. The A domain presents high homology with hornerin repeats and the B domain is homologous to FLG [62][5,59]. FLG2 is also expressed in the keratohyalin granules in the granular layer [62][5,59]. The expression of FLG and FLG2 is downregulated in skin treated with 5% or 10% lactic acid, with such downregulation often used to define sensitive skin [63][60]. The expression of FLG and FLG2 has also been reported to be downregulated by tape stripping [64][61]. In a three-dimensional reconstituted human epidermis model, FLG2 downregulation was found to induce parakeratosis, compact stratum corneum, increased pH, and reduced amounts of free amino acids with reduced proteolytic processing of corneodesmosin, hornerin, and filaggrin in parallel with reduced amounts of caspase-14 [65][62]. In another report, the expression of FLG2 was described as being colocalized with corneodesmosin in the cornified cells [66][63]. The absence of FLG2 induces the marked reduction of corneodesmosin expression [66][63]. Thus, FLG2 exerts a specialized function different from FLG.
Under physiological conditions, IVL is detected from the uppermost spinous layer to granular layer keratinocytes (early-phase epidermal terminal differentiation), but the expression of LOR, FLG, and FLG2 is more confined to granular cell layer keratinocytes (late-phase epidermal terminal differentiation) [5,64–66].
3. Medicinal Use of AHR/NRF2 Dual Activators for AD
- Medicinal Use of AHR/NRF2 Dual Activators for AD
Apart from long-lasting and hazardous AHR ligands, many phytochemical AHR ligands play potentially salubrious roles for skin barrier function by upregulating FLG, LOR, and IVL [21–25]. Some AHR ligands are potent NRF2 activators and are expected to be valuable for medicinal use for eczematous skin diseases [67][21,147] where oxidative stress is upregulated [68][69][148,149]. Antioxidative AHR agonists such as coal tar [31], soybean tar glyteer ,[25,26], and tapinarof [70][71][72][150–152] have been widely used or undergone clinical trials for the treatment of inflammatory skin diseases including AD (Figure 2). Coal tar has been integrated into topical skin treatments for more than 2000 years [31]. Glyteer is a delipidated soybean tar licensed in 1924 in Japan and is still covered under the Japanese national medical insurance system as an ointment, in which it is mixed with dexamethasone [73][25,153]. Tapinarof {5-[(E)-2-phenylethenyl]-2-[propan-2-yl] benzene-1,3-diol, WBI-1001, GSK2894512 or benvitimod} is a naturally derived (but now fully synthetic) hydroxylated stilbene produced by bacterial symbionts of entomopathogenic nematodes [74][75][150,152,154,155]. Coal tar, glyteer, and tapinarof could feasibly upregulate the EDC molecules, including FLG, IVL, and hornerin, via AHR activation and exert their antioxidative function via NRF2 activation [25,31,152]. Recent clinical trials of topical tapinarof have proved its efficacy for AD compared with placebo control [76][150,151,156].
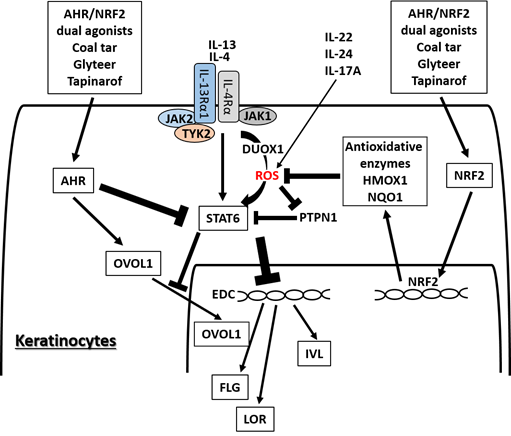
Figure 2. Medicinal AHR/NRF2 dual agonists can restore the IL-4/IL-13-STAT6-induced downregulation of FLG, LOR, and IVL expression. Coal tar and glyteer activate AHR and inhibit the STAT6 effects partially by increasing the entry of OVOL1 into the nucleus. IL-4 and IL-13 activate dual oxidase protein 1 (DUOX1), generate reactive oxygen species (ROS) production, and promote STAT6 phosphorylation in keratinocytes. On the other hand, the activation or phosphorylation of STAT6 by IL-4 and IL-13 is negatively regulated by protein-tyrosine phosphatase, nonreceptor-type 1 (PTPN1) because PTPN1 dephosphorylates the phosphorylated STAT6. ROS induced by IL-4 and IL-13 inhibit PTPN1 activity and subsequently enhance STAT6 phosphorylation. AHR/NRF2 dual agonists also activate NRF2 and upregulate the antioxidative enzymes such as NAD(P)H quinone oxidoreductase 1 (NQO1) and heme oxygenase 1 (HMOX1), which neutralize the IL-4/IL-13-induced ROS and downregulate STAT6 phosphorylation by revitalizing the PTPN1 activity. IL-22, IL-24, and IL-17A also induce ROS production.
AHR and NRF2 dual activators, coal tar and glyteer, have been shown to restore the IL-4/IL-13-mediated downregulation of IVL, LOR, and FLG [25,31]. IL-4 and IL-13 activate dual oxidase protein 1 (DUOX1), generate ROS production and promote STAT6 phosphorylation in keratinocytes [77][157] (Figure 4). On the other hand, the activation or phosphorylation of STAT6 by IL-4 and IL-13 is negatively regulated by protein-tyrosine phosphatase, nonreceptor-type 1 (PTPN1) because PTPN1 dephosphorylates the phosphorylated STAT6 [77][78][79][157–159]. Oxidative stress induced by IL-4 and IL-13 inhibits PTPN1 activity and subsequently enhances STAT6 phosphorylation [77][78][79][157–159]. Coal tar activates NRF2 and upregulates antioxidative enzymes such as NAD(P)H quinone oxidoreductase 1 (NQO1), which neutralize the IL-4/IL-13-induced ROS and downregulate STAT6 phosphorylation by revitalizing the PTPN1 activity [31]. The antioxidative AHR agonist glyteer inhibits the IL-4-induced downregulation of FLG, and activates NRF2 and downstream antioxidative enzymes such as NQO1 [24,25]. NRF2 activation also upregulates various other antioxidative enzymes such as heme oxygenase 1 (HMOX1) [80][40,42,43,143,160] and glutathione peroxidase 2 (GPX2) [81][161]. As oxidative stress is also capable of enhancing STAT3 activation, it is possible that antioxidative agents may inhibit the STAT3 pathway [82][83][162,163].
In parallel with this, other NRF2-activating phytochemicals exhibit similar inhibitory action on IL-4/IL-13-mediated FLG downregulation [20,24], IL-13-mediated periostin upregulation [143] and imiquimod-induced STAT3 activation [84][85][164,165] in keratinocytes. IL-4 also stimulates dendritic cells via STAT6 activation to produce CCL17 and CCL22, which are potent chemokines for recruiting Th2 cells [86][166]. In addition, IL-4 stimulates dendritic cells to upregulate the expression of receptors for IL-31, which is the major pruritogenic cytokine in AD [87][167]. In addition, antioxidative glyteer inhibits the IL-4/STAT6-mediated expression of CCL17 and CCL22 expression as well as upregulation of the IL-31 receptor [86][87][166,167].
OVOL1 is an important transcription factor for epidermal terminal differentiation. It resides in the cytoplasm in a steady-state condition, but activated OVOL1 translocates into the nucleus and regulates downstream gene expression [26,90,92]. OVOL1 suppresses c-Myc expression and inhibits keratinocyte proliferation ,[93,94], but it conversely promotes epidermal differentiation and upregulates the expression of FLG and LOR [22,26,90]. AHR activation upregulates the expression of OVOL1, induces its cytoplasmic-to-nuclear translocation, and increases the expression of FLG and LOR [22,26,90,92]. IL-4 does not affect or rather enhances OVOL1 expression, but it inhibits the cytoplasmic-to-nuclear translocation of OVOL1, which correlates with the downregulation of FLG expression [26,90]. AHR activation restores the IL-4-mediated inhibition of OVOL1 nuclear translocation and recovers the IL-4-induced FLG downregulation [26,90]. Interestingly, AHR activation also upregulates IVL expression, but its regulation is OVOL1-independent [22]. In addition, IL-4 and IL-13 themselves increase the mRNA and protein expression of AHR in B cells and keratinocytes [88][89][168,169]. This suggests a mutually compensatory (or seesaw) regulation between Th2 and AHR signaling. Keratinocytes are a rich source of pro-Th2 cytokines such as IL-33 [98]. AHR-mediated OVOL1 activation is also functional in inhibiting IL-33 production in keratinocytes [90][170].