Hyperoxia effects on cancer biology were explored following multiple pathways, using both in vitro cancer cell cultures and in vivo tumoral animal models. Experimental data support clinical evidences demonstrating that hyperoxia, mainly if prolonged, can induce lung injury and cerebral damage, and this can be counteracted by down-modulation of Akt, by low-dose vitamin D or aspirin.
1. Background
Despite the development of a more targeted and personalised multimodal treatment, cancer mortality remains high. In the USA, during 2020, a COVID-19 pandemic year, cancer was responsible for almost 600,000 casualties, representing the second leading cause of death after heart disease [1][2]. The main cause of death is cancer progression, the occurrence of distal metastasis or cancer recurrence, as the vast majority of patients are now successfully treated for primary tumours, with 64% of them having more than 5 years survival [2][3].
In cancer patients receiving general anaesthesia for surgery, increased inspiratory oxygen concentration is commonly used both intraoperatively and, for a variable time period, postoperatively, in order to prevent the potential development of hypoxemia and to ensure an adequate oxygen transport to the organs, tissues and cells. For a long time considered as a useful and harmless intervention, oxygen therapy has been under scrutiny in recent years due to the accumulation of data regarding its deleterious effects in critically ill patients [3][14] and many areas of acute medicine (stroke, acute myocardial infarction, cardiac arrest) [4][5][6][7][8][15,16,17,18,19]. In critically ill patient care, current data does not support a benefit from supranormal oxygen delivery. A meta-analysis of 19 clinical studies showed that hyperoxaemia was associated with increased in-hospital mortality [9][20]. A large observational multicentric study, collecting almost 300,000 arterial blood gas analysis from more than 14,000 critically ill patients, demonstrated a linear relationship between hyperoxemia and mortality, with both duration and severity adversely affecting the outcome [10][21]. A recent systematic review of 16,000 critically ill patients suggested that hyperoxia exposure might be associated with increased hospital mortality [11][22]. The British clinical practice guideline for oxygen therapy in acutely ill medical patients recommends to stop supplemental oxygen therapy when SpO 2 reaches 96% and not to start oxygen therapy at or above 93% oxygen saturation in patients with acute stroke or myocardial infarction (strong recommendations) [12][23].
During the perioperative care of an oncologic patient, the anaesthetist may currently choose between different types of anaesthesia techniques (general anaesthesia versus regional or neuraxial anaesthesia) and can also choose between various anaesthetic and analgesic agents (inhalation versus intravenous anaesthesia). However, oxygen, the most commonly used perioperative drug, cannot be replaced. The optimal dose and duration for oxygen therapy in surgical cancer patients is not clearly established.
2. Potential Molecular Mechanisms Exploring Hyperoxia Effects on Cancer Progression
2.1. ROS Production and Oxidative Stress
Hyperoxia never occurs during natural circumstances. Thus, while normal tissues and cells have extensive adaptive mechanisms to hypoxemia, they have limited protection against hyperoxia. In order to maintain normal cellular functions, a highly regulated balance between oxidant and antioxidant molecular activity emerged during evolution. High concentration oxygen exposure results in increased ROS formation and oxidative stress, overwhelming the antioxidant mechanisms capacity and producing DNA damage, protein damage and lipid peroxidation
[13][70]. ROS are constantly generated and eliminated in biological systems via several regulatory pathways. ROS production is modulated by endogenous and exogenous factors, including oxygen, environmental stressors or ionizing radiation in a dose dependent manner. While intrinsic levels of ROS are involved in the maintenance of cellular homeostasis, high levels of ROS can be toxic to cells. Thus, oxidative stress may lead to cell damage or cell death by apoptosis or necrosis. The severity of cell disturbances depends on the cumulative oxygen dose (concentration and duration of exposure) and on cell susceptibility. At the same time, when a cell experiences high stress levels, it activates autocrine and paracrine mechanisms to protect itself and other cells. In clinical practice, these subtle effects are almost silent and undetected during the postsurgical recovery but may have long-term outcome consequences.
In surgical patients, a recent study confirmed that intraoperative and postoperative hyperoxia exposure (administration of FiO
2 = 0.80 in anesthetized patients undergoing abdominal surgery) alters the redox equilibrium at 24 h after surgery, demonstrated by increased lipid peroxidation and decreased antioxidant barrier strength
[14][71].
In many human solid cancers, an important difference in oxygen concentration between the peripheral and the central tumoral cells has been described
[15][16][72,73]. This variation, mainly explained by the rapid tumoral growth related to the peripheral distribution of blood vessels and abnormal angiogenesis, leads to a hypoxic environment inside the tumoral core. The molecular mechanisms triggered by tumoral hypoxia have been widely investigated. The decreased oxygen availability (intra-tumour hypoxia) will trigger an adaptive response, initially represented by up-regulation and increased levels of the Hypoxia Inducible Factor-1α (HIF-1α) protein. In addition, major genetic and epigenetic alterations already present on the cancer cells can further increase HIF-1α activity
[15][72]. HIF-1α accumulates and induces, at the nuclear level, the transcription of multiple target genes. This process is followed by the synthesis of different proteins involved in angiogenesis, such as Vascular endothelial growth factor A (VEGF-A), in metabolic shift, such as glucose transporters GLUT-1, GLUT-3, in pH adaptation, such as carbonic anhydrase enzymes CA-9, CA-12, and also in migration, invasion, proliferation and metastasis, such as Insulin-like growth factor (IGF-2) and E-Cadherin
[16][73].
Various regimens of normobaric or hyperbaric high oxygen concentrations are used to re-sensitize chemoresistant cancer cells
[17][18][74,75], as a treatment of various cancer types
[19][20][21][76,77,78], as a modulator of immune system anti-cancer responses
[22][79] or in wound healing and neuroprotection
[23][24][25][80,81,82].
2.2. Hyperoxia and the Immune System
Hypoxia has the ability to regulate immunosuppressive mechanisms that involve myeloid-derived suppressor cells (MDSCs), tumour-associated macrophages (TAMs), regulatory T-cells (Treg cells) and immune checkpoint pathways, such as the programmed cell death-1 (PD-L1). Using a murine model of triple negative breast cancer, Qian et al. demonstrated that long-time respiratory hyperoxia exposure, 60% O
2 concentration administered continuously for 21 days can reverse immunosuppression by regulating MDSCs and PD-L1 expression
[22][79]. Moreover, Hatfield et al. demonstrated, in a murine model of lung cancer, that increased inspiratory oxygen concentrations decreased immunosuppressive molecules, such as transforming growth factor–β (TGF-β), weakened immunosuppression by regulatory T cells and improved lung tumour regression and long-term survival in mice
[26][83]. In a non-cancer model, Kumar et al. demonstrated that neonatal hyperoxia alters the adaptive immune response in adult mice, a mechanism which increase the risk for susceptibility to infection in premature infants
[27][84].
The interaction of hyperoxia with the immune system will require additional investigations regarding the oxygen concentration, exposure length and patient status as there is another piece of evidence that indicates that hyperoxia does not exert immunologic effects in murine and human experimental setups
[28][85]. The exposure of mice for 2.5 h or of healthy volunteers for 3.5 h to 100% oxygen does not affect the inflammatory response induced by administration of endotoxin. The data suggest that short-term hyperoxia has different effects on cancer patients compared with healthy volunteers.
2.3. Angiogenesis and Epithelial Mesenchymal Transition (EMT)
In an in vitro experimental study testing xenon- and sevoflurane-mediated effects on the migration and expression of angiogenesis biomarkers in human breast adenocarcinoma cells, Ash et al. have also documented increased migration of breast cancer cells, when exposed to 65% oxygen as compared to 25% oxygen
[29][86]. Crowley et al. tested the effects of four oxygen concentrations, 21%, 30%, 60% and 80%, O
2, on both ER+ and ER− breast cancer cell lines, and found that, short-term, 3 h of 60% oxygen exposure enhances migration and also promotes the secretion of several pro-metastatic angiogenesis factors such as VEGF, IL-8 and angiogenin
[30][87].
Epithelial mesenchymal transition (EMT) is an essential cellular program, normally active during embryogenesis and wound healing. During this biological process, a normal epithelial cell, in contact with the basement membrane, turns into a mesenchymal phenotype and migrates from the original epithelial layer. The onset of EMT in cancer was associated with an increased risk of metastatic disease and cancer progression
[31][32][88,89]. EMT is accompanied by increasing the expression of the mesenchymal markers (e.g., vimentin) and by decreasing the expression of the epithelial markers, such us E-Cadherin. However, in partial EMT, cells may co-express both mesenchymal and epithelial markers or may lose epithelial markers without gaining mesenchymal markers. Partial EMT plays an important role in metastasis by enhancing tumour cell plasticity
[32][89]. The increased expression of Vimentin, a cytoskeleton protein associated with a migration phenotype, was found to be a poor prognostic marker in cancer
[33][34][90,91]. The reduced expression of E-Cadherin, a cytoplasmic protein present in the epithelial cells, correlates, in an experimental setting, with an invasive phenotype
[35][92]. Tiron et al. demonstrated that in vitro exposure of ER-breast cancer cell lines to 80% O
2 for 6 h increases ROS and induces BDNF, VEGF-R2 and vimentin expression and thus promotes EMT and angiogenesis
[36][93]. Tested in vivo, in a murine model of ER-breast cancer, the perioperative exposure to 80% oxygen for 6 h was associated with an increased number and size of hepatic metastasis, evaluated at 4 weeks after the surgical excision of the tumour
[36][93].
2.4. Brain-Derived Neurotrophic Factor (BDNF)
Hyperoxia increases ROS and activates cell defence systems in order to keep ROS within physiological range, as previously discussed. BDNF is one of the molecules that are upregulated in response to hyperoxia exposure, as shown in various experimental settings, including the peribronchial smooth muscle of neonatal rats
[37][95], the Alzheimer mouse model
[38][96], the breast cancer mouse model
[36][93] and healthy volunteers
[39][97]. Interestingly, some healthy volunteers enrolled in the study
[39][97] resigned due to the side effects, although the tested oxygen concentration was only 37% O
2. BDNF and its main receptor, tropomyosin receptor kinase B (TrkB) have been reported to promote alveolar epithelial regeneration after lung injury
[40][98] or to exert neuroprotective effects
[41][42][43][44][45][46][99,100,101,102,103,104]. However, TrkB can be activated by cyclic adenosine monophosphate
[47][105] in order to exert the neuroprotective effects. These data suggest that TrkB plays a pivotal role in response to ROS. However, the activation signals can come either from BDNF or from other agonists, depending on cell type and ROS sub-type. The interaction BDNF-ROS does not consist only in BDNF overexpression in response to increasing levels of ROS, as it has been reported that BDNF exerts various biological effects by the generation of oxidative stress in human vascular endothelial cells
[48][49][106,107].
BDNF-TrkB axis exerts various effects depending on the cell types. In normal cells, it protects cells from high levels of ROS or induces moderate ROS levels in other circumstances. In cancer cells, in addition to pro-survival signalling, the BDNF-TrkB molecular pathway induces EMT, which is associated with poor prognostic in various cancer types
[50][51][108,109], may increase migration and invasion
[52][53][110,111], may promote cancer cell survival and proliferation
[54][55][112,113] and may increase neo-angiogenesis through increasing vascular endothelial growth factor (VEGF) expression
[56][57][114,115]. The authors from
[58][116] reported that BDNF-TrkB axis induces VEGF expression via HIF-1α. HIF-1α is generally accepted to have an increased expression in hypoxia and a decreased expression in hyperoxia conditions. Tiron et al. identified an upregulation of HIF-1α protein expression after short hyperoxic exposure of triple negative breast cancer cells in an experimental mouse model of perioperative care
[36][93]. These data join the new sparse data that support HIF-1α increased protein expression after the exposure to hyperoxic episodes
[23][59][80,117] and suggest that, in certain circumstances (e.g., mild hyperoxia), HIF-1α protein expression is not impaired as in prolonged high grade hyperoxia. In non-cancer patients, HIF-1α may have beneficial effects (e.g., muscle regeneration), but in oncologic patients it may promote cancer progression and recurrence.
Cells that are detaching by the extracellular matrix will undergo a process of cell death called anoikis. There are data
[60][61][62][63][118,119,120,121] demonstrating that BDNF-TrkB axis induce anoikis resistance in various cancer types, in addition to EMT. BDNF synthesized as the result of perioperative oxygen exposure in oncological patients may promote survival of the circulating tumour cells and formation of new metastases; in addition it can decrease the time to relapse by increasing cancer cell proliferation and EMT at the level of existing micrometastases.
Chemoresistance occurs in many types of cancer in clinical setting. In breast cancer, it has been reported that multi-nucleated cells generated by chemotherapy drugs are oxidatively stressed, and this induces chemoresistance, as shown both in vitro and in vivo
[64][122]. Moreover, the multi-nucleated cells induce chemoresistance by secreting VEGF, activating the RAS/MAPK pathway and, thus, ROS—HIF-1α signalling plays a crucial role. It could be possible that oxidatively stressed multi-nucleated cells to increase the expression of BDNF in order to counteract ROS, and BDNF would be responsible for activating the HIF-1α-VEGF pathway, which further induces chemoresistance.
Another source of ROS production in cancer is represented by ionizing radiation. Cancer radiotherapy changes the tumour microenvironment, which in some circumstances induces resistance and recurrence, as already published
[65][66][123,124]. BDNF does not emerge as a key molecule in radio-resistance; however, the classical downstream signalling pathway of the receptor tyrosine kinase family is involved, as inhibition of PI3K/mTOR induced radio-sensitization in pediatric and adult glioblastoma
[67][125]. However, it has been shown that low-level laser therapy can rescue dendrite atrophy by upregulating BDNF expression via ROS generation
[68][126]. These data suggest that, in certain cancer types, subtypes and radiation clinical models, cancer cells are able to express enough BDNF to protect themselves against the increased ROS levels generated by radiation, the mechanism responsible in many cases for radio-resistance phenomena. On the other hand, it has been demonstrated that total abdominal irradiation causes cognitive deficits in mice models
[69][127]. Abdominal irradiation shifted gut bacterial composition, which increased the expression of miR-34a-5p in small intestine tissue and also in the peripheral blood. Expressed miR-34a-5p targeted the 3′UTR of BDNF mRNA in hippocampus to mediate cognitive dysfunction. This mechanism was further prevented by tail intravenous injection of a miR-34a-5p antagomir. All together, these data may explain why the irradiation of some colon cancers is associated with good prognosis while, in other cases, radio-resistance occurs. Increased BDNF levels may favour radio-resistance by its pro-survival role and also by the induction of the epithelial to mesenchymal transition (EMT) process. On the other hand, a decreased BDNF level induced by irradiation and the shift in the bacterial composition of gut microbiota may sensitize the cancer cells.
2.5. Hyperoxic-Hypoxic Paradox
In clinical circumstances, fluctuations in oxygen levels due to hyperoxic episodes may induce the expression of many mediators usually induced by hypoxia. This phenomenon, known as the “hyperoxic-hypoxic paradox” has been recently reviewed by Hadanny and Efrati
[70][128]. Although the hyperoxic-hypoxic paradox was identified after repeated intermittent hyperoxia, Tiron et al.
[36][93] demonstrates the expression of HIF-1α and VGFR after a single hyperoxia exposure, factors which are normally expressed under hypoxic conditions.
While immunosuppression mediated by tumour hypoxia has been extensively investigated, the data regarding the interaction of perioperative hyperoxia with immune system is lacking. HIF-1α plays an important role in immunosuppression, mediated by chronic hypoxia. We speculate that HIF-1α expressed after acute perioperative hyperoxia may interact with the immune system, probably in a lower extend compared with chronic hypoxia.
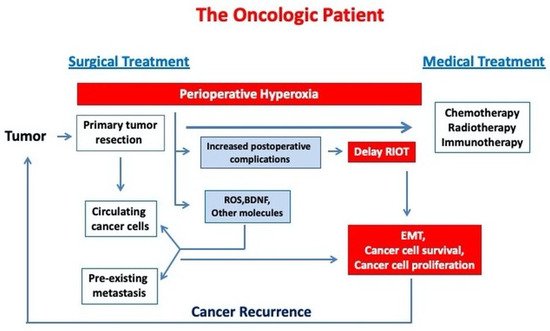
Figure 1 describes the link between perioperative hyperoxia exposure and cancer progression in oncologic patients.
Figure 1. Perioperative hyperoxia may induce cancer progression in oncological patients by increasing cancer cell survival and EMT. (RIOT—return to the intended oncological treatment, EMT—epithelial mesenchymal transition).