Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Camila Xu and Version 4 by Camila Xu.
The genus Burkholderia belongs to the subphylum of β-proteobacteria and encompasses Gram-negative bacterial species with high genetic versatility and adaptability to various ecological niches.
- Burkholderia genomics
- phytopathogenic Burkholderia
- plant symbiotic Burkholderia
1. Introduction
The genus Burkholderia belongs to the subphylum of β-proteobacteria and encompasses Gram-negative bacterial species with high genetic versatility and adaptability to various ecological niches [1]. More specifically, they are able to occupy ecosystems as diverse as soils, plants, and even animal and human bodies as serious disease-causing agents [2][3][4][2,3,4]. Recently, Burkholderia organisms have drawn increasing attention as abundant and fundamental components of various ecosystems. The first member of the genus was isolated in 1942 from carnations displaying wilt and root-rot symptoms. It was initially named Phytomonas caryophylli and later renamed Pseudomonas caryophylli [5]. In 1950, another species was isolated from onions showing sour skin rot and named Pseudomonas cepacia [6][7][6,7]. Some members, originally included in the genus Pseudomonas, were later reclassified in the new Burkholderia genus due to improved taxonomic tools, the availability of techniques for DNA–DNA hybridization, as well as for 16S rRNA analysis [8]. Since then, many other species have been, and are still being, described and added to the genus.
The adaptability of Burkholderia spp. to diverse environments and their ecological versatility rely on the high genetic plasticity of their multi-chromosome genomes, rich in insertion sequences [9] and exhibiting a relatively high proportion of coding regions, allowing these bacteria to produce various metabolites with degradative potential [10]. Such metabolic capacity is a fundamental feature for their survival and fitness in different habitats [11].
The Burkholderia cepacia complex (BCC), one of the major components of the Burkholderia genus, comprises a group of related opportunistic human pathogen species responsible for pulmonary infections, particularly in immunocompromised and cystic fibrosis patients [9]. Due to the importance of BCC members as human pathogens, intensive research efforts and discussion have been devoted to the epidemiology of human diseases caused by Burkholderia spp. Other related species include animal pathogens such as Burkholderia mallei and plant pathogens such as Burkholderia glumae, Burkholderia gladioli, and Burkholderia plantarii. Notably, another group of plant-associated Burkholderia spp. comprises potentially beneficial species of plant growth-promoting or plant-symbiotic bacteria, favoring the healthy growth of plants by nitrogen fixation and protection against plant diseases. From the taxonomic standpoint, an effort is being made to better discriminate beneficial from pathogenic Burkholderia spp. [12].
2. Taxonomic Updates of the Burkholderia Sensu Lato
Following the proposal of Burkholderia as a new genus in 1992 [8], and the continuous increase in the number of species within it, several taxonomic rearrangements have been suggested. Pieces of evidence from 16S rRNA analysis and the construction of phylogenetic trees based on other genes, such as recA, have increasingly supported the division of the Burkholderia genus into two distinct lineages [13]. Based on the suggested division, one group comprises human, animal, and plant pathogens or opportunistic pathogens, whereas the other group is represented by environmental xenobiotic-biodegrading, plant symbiotic, and plant growth-promoting species.
The distinction between the two lineages has been further supported by the identification of genomic within-group similarities and between-group differences. Ussery et al. [14] explored the genomic diversity among members of the Burkholderia genus and defined the pan- and core-genomes using the genome sequences available at the time of that study (56 full and partial genome sequences). A phylogenetic analysis conducted on 612 genes from the core Burkholderia genome resulted in a clear separation of the pathogenic Burkholderia spp. from members of the other clade. By combining comparative genomics with phylogenetic analyses based on 16s rRNA, recA, gyrB, and acdS genes, Gyaneshwar et al. [15] suggested the distinction of the Burkholderia genus into two subgenera and initially proposed the new genus, Caballeronia.
Consistently, multilocus sequence analysis using four housekeeping genes, namely atpD, gltB, lepA, and recA, in combination with 16S rRNA, confirmed the presence of different Burkholderia lineages by separating the genus into two major clusters [16]. The distinction was even more obvious after bioinformatics analysis of the virulence loci of Burkholderia genomes and functional pathogenicity tests [17]. In light of compelling evidence on the existence of systematic differences within the Burkholderia group, a novel genus, Paraburkholderia, was proposed based on the analysis of conserved sequence insertions/deletions [18]. Since then, several other species have been ascribed to the Paraburkholderia genus and the previously proposed Caballeronia genus [19]. Following reassessment of Burkholderia andropogonis, this species was separated into a new genus, Robbsia, as confirmed by phylogenetic and comparative genomic analysis [20]. Using whole-genome sequence data, Beukes et al. [21] addressed these complex phylogenetic relationships and confirmed the existence of generic boundaries among Burkholderia sensu lato bacteria. Although the analyzed genomes were similar in size and number of encoded genes, the genomes of Burkholderia sensu stricto showed higher G+C contents. The latter study includes a large-scale phylogenetic analysis on 106 common genes of Burkholderia sensu lato, specifically selected to limit phylogenetically irrelevant data. The results support the subdivision of Burkholderia sensu lato into five distinct lineages: Burkholderia sensu stricto, Paraburkholderia, Caballeronia, Robbsia, and Paraburkholderia rhizoxinica [21]. More recently, whole-genome analysis has been employed in the context of an international joint effort to investigate the taxonomic status of selected species of Burkholderia sensu lato [22]. Based on the phylogenetic analysis of conserved genes and average nucleotide and amino acid identities, two novel genera were proposed within the Burkholderia sensu lato assemblage. The first, Mycetohabitans, encompasses fungal symbiotic species with relatively small genomes. The second, Trinickia, includes soil and plant-associated species. Furthermore, the comparison of core-genomes revealed the presence of functional genus-specific genes. A historical overview of the major rearrangements and updates in Burkholderia taxonomy is summarized in Table 1.
Table 1.
An overview of the major rearrangements and updates in the taxonomy of
Burkholderia
.
| Year | Finding | Details | Reference |
|---|---|---|---|
| 1942 | First isolation of Burkholderia | Originally named Phytomonas caryophylli; then Pseudomonas caryophylli | [5] |
| 1992 | A new Burkholderia genus was proposed | The new genus comprised seven species from the genus Pseudomonas | [8] |
| 2011 | A second genus (Caballeronia) was suggested | Based on phylogenetic analysis of multiple genes and comparative genomics; however, the evidence was not sufficient to confirm the new grouping | [15] |
| 2014 | The genus Paraburkholderia was proposed | Based on the analysis of conserved sequence in/dels | [18] |
| 2016 | Inclusion of several species in the Paraburkholderia genus and establishment of the Caballeronia genus | Eleven species were reclassified as Paraburkholderia and 14 species were transferred to the newly established Caballeronia genus | [19] |
| 2017 | Burkholderia andropogonis was separated in a newly proposed genus as Robbsia andropogonis | Based on multilocus sequence, 16S rRNA gene phylogeny, and average nucleotide identity analyses, as well as tetranucleotide signature frequency and percentage of conserved proteins | [20] |
| 2017 | Confirmation of the genetic boundaries among the 4 established groups and suggestion of a fifth division: Paraburkholderia rhizoxinica | Five groups (Burkholderia sensu stricto, Paraburkholderia, Caballeronia, Robbsia, Paraburkholderia rhizoxinica) were separated based on maximum likelihood phylogenies using the amino acid and nucleotide sequence of 106 conserved proteins | [21] |
| 2018 | Two novel genera (Mycetohabitans and Trinickia) were proposed | Based on whole-genome comparative study and phylogenetic analysis of conserved genes | [22] |
It is clear that the Burkholderia sensu lato group has undergone several taxonomic changes and could be subjected to further adjustments in the future. The increased availability of genomic information, as well as the advancements in bioinformatics analysis, have enhanced our understanding of the evolutionary and taxonomic relationships between clinically, environmentally, industrially, and agriculturally important Burkholderia species. All this considered, the large group of Burkholderia sensu lato is currently composed of six genera, i.e., Burkholderia sensu stricto, Caballeronia, Paraburkholderia, Robbsia, Mycetohabitans, and Trinickia. The six genera were established after phylogenetic analysis of the main representative Burkholderia sensu lato strains using the neighbor-joining method, based on 16S rRNA sequences retrieved from the SILVA database (https://www.arb-silva.de/), as shown in Figure 1.
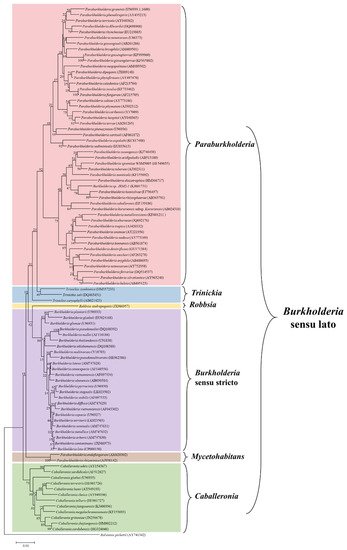
Figure 1. Phylogenetic tree constructed by the neighbor-joining method using 16S rRNA sequences of representative strains of Burkholderia sensu lato assemblage. The 16S rRNA sequences were retrieved from the SILVA database (https://www.arb-silva.de/). Species names were corrected to match the current taxonomic positions and the accession numbers for each sequence are given in parenthesis following the species name. The updated genera and their phylogenetic relationships are also shown. Ralstonia pickettii (AY741342) was used as an outgroup.
With regard to the scope of the current review, phytopathogenic Burkholderia species are classified in the Burkholderia sensu stricto group, closely related to the Burkholderia cepacia complex; the former Burkholderia andropogonis is currently a separate genus as Robbsia andropogonis; and Burkholderia caryophylli is now classified as Trinickia caryophylli. On the other hand, most of the plant-beneficial Burkholderia species were transferred to the Paraburkholderia and Caballeronia genera.
3. Genomic Features of the Plant-Associated Pathogenic and Beneficial Burkholderia
Because of the clinical importance of the B. cepacia complex, special consideration is devoted to these bacteria by genomic studies and, therefore, their genomes are well-characterized [9]. Besides common genomic features shared by most organisms of the large Burkholderia sensu lato group, functional features specific to the plant-associated groups are present and allow them to occupy various ecological niches with different lifestyles, as pathogens, legume nodulators, or biocontrol agents. Common features of Burkholderia species include relatively large multi-replicon genomes, the presence of genomic islands, and multiple insertion sequences conferring high genome plasticity [9][10][9,10].
3.1. Genome Size
Generally, Burkholderia organisms have larger genomes (7–8 Mbp on average) than other bacteria; however, the genome size greatly varies among different Burkholderia groups. The smallest genome among the free living Burkholderia (3.75 Mbp) is found in the fungal endosymbiont, Mycetohabitans rhizoxinica (basonym Burkholderia rhizoxinica), whereas the largest genome (11.5 Mbp) is found in the soil bacterium Paraburkholderia terrae (basonym Burkholderia terrae). It is suggested that genome size can provide insights into the evolutionary history and the relationships between Burkholderia species. It is also noted that the particular lifestyle of Burkholderia species might reflect selective pressure for a specific genome size [23].
Two main models for genome evolution in Burkholderia species were proposed: (i) plasmids might undergo rearrangements following parallel transfer, becoming established as additional chromosomes or megaplasmids and allowing for bacterial acquisition of essential genes; and (ii) loss of genomic information following rearrangements [24]. An example of the latter mechanism could be M. rhizoxinica, the obligate endosymbiont of the zygomycete Rhizopus microsporus, which is a causal agent of rice seedling blight development by production of rhizoxin [25]. The genome of M. rhizoxinica, containing one chromosome, a megaplasmid, and a plasmid, is remarkably small (relative to other Burkholderia), most likely because of rearrangements and deletion of genomic information, especially in genes coding for mobile genetic elements. The endosymbiotic lifestyle reduced the bacterial capacity to adapt to varying habitats and made efficient sensing superfluous [26][27][26,27]. On the other hand, a large part of the chromosomal coding region is devoted to the production of the secondary metabolite, rhizoxin [26][27][26,27].
Regarding the plant symbiotic Burkholderia, which can also be free-living in the bulk soil or water, a characteristic large genome size could explain their versatile lifestyle and the capacity to perform many plant-beneficial functions [28].
In another comparative genomic study of phytopathogenic Burkholderia species, Seo et al. [29] analyzed the genome sequences of different strains of B. glumae, B. gladioli, and B. plantariiand found genome size variations not only between species but also between different strains of the same species. For B. glumae, the smallest genome size, ~4.9 Mbp, was exhibited by the AU6208 strain, whereas the largest, ~7.2 Mbp, was found in the BGR1 strain. The relatively small genome size of B. glumae AU6208 could be attributed to genome rearrangements or deletions, as a result of adaptation to different hosts. Notably, although pathogenic to rice, AU6208 was originally isolated from infant patients, unlike other strains that were all isolated from rice as the original host. The genome size of B. gladioli BSR3 is ~9 Mbp and that of B. plantarii is ~8 Mbp.
3.2. Multi-Replicon Nature
A multi-replicon genome structure is found in most Burkholderia species with various numbers of chromosomes and plasmids. Martínez-Aguilar et al. [28] studied the multi-replicon genomes of plant-associated diazotrophic Burkholderia species and found that three strains of Paraburkholderia unamae and Paraburkholderia silvatlantica (formerly Burkholderia spp.) contain genomes of four replicons. The largest and smallest replicon sizes are ~3.31 and ~1.11 Mbp, respectively. In addition, the genomes of three strains of Paraburkholderia tropica (formerly Burkholderia tropica) contain five replicons ranging from ~3.24 to ~0.53 Mbp. The authors suggested that such multi-replicon large genomes could explain the capacity of these diazotrophic Burkholderia species to efficiently colonize the rhizosphere as well as endophytic environments.
A multi-replicon genome structure is also reported among the plant-pathogenic Burkholderia species. The genomes of the representative strains B. glumae BGR1 and B. gladioli BSR3 contain two chromosomes and four plasmids, while the B. plantarii ATCC43733 genome contain two chromosomes and one plasmid [29]. Agnoli et al. [30] found that deletion of the third chromosome from the genomes of several BCC species results in the attenuation of virulence and the loss of many functions such as the ability to utilize several substrates, antifungal activity, and biosynthesis of extracellular polymeric substances. Intriguingly, bacterial viability is not significantly affected, despite the massive loss of genomic information (~1 Mbp). This finding indicates that multi-replicon genomes are essential for adaptation but not necessarily for bacterial growth and survival.
While studying the pan- and core-genomes of Burkholderia species, Ussery et al. [14] found that only few hundred genes are conserved among all genomes to form the Burkholderia core genome, whereas more than 40,000 gene families are present in the Burkholderia pan genome. A more recent study of the available genome sequences, focusing on 110 genome sequences, shows that around 35,680 genes belong to pan-gene and 717 to core-gene families [23]. In the latter study, the number of shared genes has significantly dropped upon adding genes from different replicons to the analysis, which indicates the low percentage of genes shared by different genomic replicons of Burkholderia. In the same context, Seo et al. [29] analyzed the genomes of 106 Burkholderia species, representing the different groups, and found that 78,782 genes correspond to pan-genomes, while 587 are core genes conserved in all tested species. By considering the genomes of several strains of three plant pathogenic Burkholderia (B. glumae, B. gladioli and B. plantarii), 12,758 pan-genome genes are present in the tested strains, while 1908 genes are found conserved as the core-genome [29].
3.3. Genomic Islands and Multiple Insertion Sequences
Along with the above-mentioned features of Burkholderia genomes, other unique features are related to the genetic plasticity of these bacteria, such as the presence of genomic islands and multiple insertion sequences. Genomic islands refer to foreign DNA regions that have been horizontally acquired and became integrated within the genomes of several Burkholderia species. They have important evolutionary roles and can specify several accessory functions [31]. Multiple approaches have been used to identify these foreign DNAs, such as the analysis of G+C content, which has been found to differ in genomic islands with respect to the rest of the genome [32][33][32,33]. Genomic islands vary in their structure and content between Burkholderia species and account for more than 10% of the genomes in most species [24]. Genomic islands linked to pathogenicity and transmission have been extensively characterized. However, the coding capacity of genomic islands is not only associated with pathogenicity and virulence but also with adaptive traits such as symbiosis and metabolism [31]. An example of this is the environmental species Paraburkholderia xenovorans (formerly Burkholderia xenovorans), in which more than 20% of total genes are a recent acquisition and mainly involved in niche adaptation [34].
The genomes of Burkholderia species also contain many diverse insertion sequences that are mainly attributed to genomic rearrangements and replicon fusion and are related to transcriptional regulation [10][24][35][36][10,24,35,36]. This finding indicates that the presence of multiple insertion sequences in Burkholderia genomes has a role in phenotypic, as well as genetic diversity, by activating and/or inactivating adjacent genetic loci. Target genes may be inactivated or, in the case of adjacent promoter-less genes, activated by the promoters provided by insertion sequences [37]. Furthermore, the presence of multiple copies of the same insertion sequence may drive genetic rearrangements by fusion of two replicons, inversion, deletion, and duplication of the intervening region [38][39][38,39]. Accordingly, the transposition of insertion sequences is a very important evolutionary mechanism for the emergence of adaptation capacity and variability in Burkholderia species. A clear example of insertion sequence-mediated genomic alterations is provided by the genome of B. mallei, which is suggested to arise from several deletions and inversions of the Burkholderia pseudomallei genome. Evidence supporting such an assumption includes the presence of insertion sequences bordering most of the breakdown points and the presence of regions from chromosome 1 of B. pseudomallei on chromosome 2 of B. mallei [40]. In the phytopathogen B. glumae, the insertion sequences IS1417, IS1418, and IS1419 are characterized and found to be prevalent among different field strains. Apparently, these sequences may have been horizontally acquired by B. glumae from other related species, as they are also found in species such as B. gladioli and B. plantarii, with IS1417 being the most prevalent [41].