Microglia play a critical role in both homeostasis and disease, displaying a wide variety in terms of density, functional markers and transcriptomic profiles along the different brain regions as well as under injury or pathological conditions, such as Alzheimer’s disease (AD). The generation of reliable models to study into a dysfunctional microglia context could provide new knowledge towards the contribution of these cells in AD. In this work, we included an overview of different microglial depletion approaches. We also reported unpublished data from our genetic microglial depletion model, Cx3cr1CreER/Csf1rflx/flx, in which we temporally controlled microglia depletion by either intraperitoneal (acute model) or oral (chronic model) tamoxifen administration. Our results reported a clear microglial repopulation, then pointing out that our model would mimic a context of microglial replacement instead of microglial dysfunction. Next, we evaluated the origin and pattern of microglial repopulation. Additionally, we also reviewed previous works assessing the effects of microglial depletion in the progression of Aβ and Tau pathologies, where controversial data are found, probably due to the heterogeneous and time-varying microglial phenotypes observed in AD. Despite that, microglial depletion represents a promising tool to assess microglial role in AD and design therapeutic strategies
- microglia
- Alzheimer’s disease
- depletion
- inflammation
1. Microglia: Micro in Size but Macro in Functions, Highly Important in Alzheimer’s Disease
Microglia, the primary immune cells of the brain, not only survey the environment for pathogens and debris, but also play other important roles in the central nervous system (CNS), providing direct sustain to neurons and supporting myelinogenesis, synaptic plasticity, and the neoformation of vessels [1][2]. These glial cells account for 10–15% of the total cells in the adult CNS in humans [3] and 5–12% in mice [4]. Microglial cells derive from myeloid progenitors of the yolk sac that at embryonic day 8.5 colonize the mouse fetal brain, and actively proliferate at early postnatal days until reaching their definitive brain density [3]. Although the number of microglial cells remains constant during lifetime in mice and humans, a rapid turnover of microglia is maintained by a balanced coupling of microglial proliferation and apoptotic death [5]. In adult life, a 28% of microglia are renewed daily, meaning the lifespan for these cells 4.2 years [6]. Spatial heterogeneity of microglia has been observed in terms of density, functional markers and transcriptomic profiles. Moreover, microglia suffer transcriptional, morphological, and functional changes during aging, injury or pathological conditions, as multiple sclerosis, Parkinson’s disease (PD), and Alzheimer’s disease (AD), among others.
In this review, we specifically focused on the role of microglia in AD. In patients, AD pathology develops along a continuum process (ATN), in which the amyloid deposition is considered the earlier event, preceding and triggering Tau pathology and neurodegeneration [7][8][9]. However, microglial role in the ATN continuum remains unsolved. Microglial activation and the loss of their homeostatic functions are considered as critical features in AD pathogenesis. Recent single-cell transcriptomic studies have identified different microglial subpopulations involved in AD [10][11][12][13][14], although the functional significance of this microglial diversity is not clearly understood. Moreover, microglial activation has been widely described in AD transgenic mice, but depending on the models, timing of pathology development and brain region, activated microglia can adopt a protective role or may acquire a cytotoxic phenotype, mediating neuronal damage. In amyloidogenic AD mouse models, a subset of activated microglial cells, named “disease-associated microglia” (DAM), cluster around amyloid plaques establishing a protective barrier [15][16]. This phenotype, characterized by the upregulation of genes involved in lysosomal, phagocytic, and lipid metabolic pathways, is ApoE- Trem2 dependent [15][16][17] and requires an oxidative metabolism [18]. Similar microglial transcriptomic profiles have also been described in several tauopathy models such as P301S and P301L mice [19][20]. However, microglial response is diverse in transgenic Tau models as, for instance, ThyTau22 mice manifest mild microglial activation, whereas P301S mice exhibit a strong microglial response [20]. Although the contribution of microglial cells to the progression and spread of pathogenic Tau species is still a matter of debate, it has recently been described that TREM2 loss of functions increases neuritic pathology and Tau spreading in amyloidogenic models [21].
Although microglial activation has been reported in several brain regions of AD patients [22][23][24], it is important to point out that, in the hippocampus, the microglial response is not as strong as reported for amyloidogenic mice and several Tau models [20][25]. Apart from the individual and regional heterogeneity, this apparent discordance between transgenic models and AD patients may be associated to the aging process itself, the main risk factor for late-onset AD, and/or to the chronic pathology of AD. Mouse models bearing familial AD mutations are frequently examined at relatively young ages compared to the elderly AD patients. Nevertheless, our results and others show that microglial activation increases with age in animals models with amyloid or Tau pathologies [20][26]. Then, other comorbidities present in AD patients as vascular deficiencies, hypertension, inflammatory diseases, obesity or diabetes mellitus could be involved in this distinct microglial response. What is more, transcriptomic studies from purified microglia showed few overlaps in differentially expressed genes during aging between humans and mice, hinting that microglia may age differently in both species [27].
In the last years, microglial depletion mouse models have provided new insights into the role of these cells in physiological and pathological conditions. Here, we review different mouse models of microglial depletion, both in health and in AD, evaluating how reliable they could be as tools to study a context of microglial dysfunction or a context of microglial renewal. We recapitulate previous published data on the main microglial depletion strategies and, importantly, we also include unpublished data from our recently developed mouse model of conditional microglia depletion ( Cx3cr1 CreER /Csf1r flx/flx ). We also comment on the origin and pattern of microglial repopulation process. Additionally, we review previous works in regards to the effects of microglial depletion and repopulation in the progression of Aβ and Tau pathologies. Outcomes are diverse and sometimes contradictory, but they open new research lines regarding the mechanisms underlying microglial proliferation and migration capabilities. Selective ablation of harmful microglia within suitable time windows and their replacement by protective microglia may be a promising therapeutic strategy for AD and other neurodegenerative diseases.
2. Pharmacological and Genetic Microglial Depletion Models
Microglial viability and proliferation depend on signaling through the colony-stimulating factor1 receptor (CSF1R) [5][28][29][30] that belongs to the type III tyrosine kinase family, and is activated by two different cytokine ligands, colony stimulating factor-1 (CSF1) and interleukin-34 (IL-34) [31][32][33]. However, Csf1r is expressed on all myeloid cells [34][35], so the signaling interference through this receptor will not only affect microglial cells, but also peripheral macrophages, probably mediating an immunosuppressive effect. As it is widely known, Csf1r knock-out (KO) mice do not reach adult stage [3][36] so the suppression of this receptor should be carried out in adulthood, either through the administration of pharmacological inhibitors or through controlled genetic systems. As previously revised, different approaches give rise to variable depletion percentages, also dependent on the dose and length of treatments [37][38].
The first pharmacological approach trying to deplete microglial populations used a bisphosphonate drug, clodronate, packed in liposomes (Clo-Lip), which is rapidly taken up by phagocytic cells inducing their apoptotic death. Clo-Lip does not cross the blood–brain barrier (BBB), and consequently, needs to be administered by either intraparenchymal or intraventricular injection. Intraparenchymal Clo-Lip injection depletes between 30 and 60% of microglia 24 to 72 h after injection, but also produces astrocytic activation, releases proinflammatory cytokine and alters blood vessel integrity (reviewed in [39]). A better pharmacological strategy for microglial elimination was achieved by highly potent CSF1R tyrosine kinase inhibitors as PLX3397 and PLX647 that, after crossing the BBB, lead to microglia depletion without consequent inflammation, cytokine storm, or BBB damage, and no negative effects on mice behavior and cognition [40]. Depending on the dose and inhibitor used, different degrees of microglia depletion were reached and maintained throughout the treatment. Additional specific CSF1R inhibitors as JNJ-40346527, GW2580 and BLZ945 are available, and different studies have shown their dose-dependent effects on microglial number and phenotype [40][41]. Recently, a new and highly specific inhibitor for CSF1R, PLX5622, has been developed, improving BBB penetrance compared to PLX3397 [42]. However, and unexpectedly, the effect of these small CSF1R inhibitors is not restricted to microglia, but also affects the whole macrophage population and hematopoiesis [43]. Moreover, all these inhibitors are not specific for CSF1R, as they also inhibit three other kinases as FLT3, PDGFR, and KIT [44] and leads to broad myelosuppression, affecting macrophages, osteoclasts, and mast cells, among other cells. Additionally, it should be considered that PLX treatments may have a detrimental effect on neurons as CSF1R signaling has been demonstrated to enhance neuronal survival [45]. Actually, Shi et al. showed that PLX3397 inhibit neurite outgrowth and mildly reduce neuron number in vitro [46]. Further experimental approaches are still necessary in order to specifically deplete microglia using these small CSF1R inhibitors without affecting other cell types or tissues.
A more selective microglial depletion, with little effects on peripheral tissues, can be achieved by genetic manipulations based on the combination of cell type specific promoters coupled to suicide genes [47]. The initial approach was based on the expression of the suicide herpes simplex virus thymidine kinase ( HSV-1 TK ) transgene under the Cd11b promoter [48]. The administration of ganciclovir to Cd11b -TK mutant mice induces apoptosis of microglia, but also of CD11b+ bone marrow cells. To avoid myelotoxicity and the consequent mouse death, it is mandatory to combine this model with a bone-marrow chimera system, or alternatively administrate ganciclovir intraventricularly. Other genetic approaches to deplete myeloid population used diphtheria toxin (DT)-based models, in which myeloid promoter-driven Cre recombinase mouse lines ( Cx3cr1 Cre ) were crossed with transgenic mice harboring genes for diphtheria toxin receptor (DTR) downstream of loxP-flanked STOP sequences. In this model, the administration of DT originated the acute cell death of all myeloid cells expressing DTR [49], although reached only short-lived depletion, less than 5 days.
On the other hand, the inducible Cx3cr1 CreER line allows the targeting of microglia in a cell-type-specific and tamoxifen inducible fashion [50]. Two main Cx3cr1 CreERT2 inducible lines were created separately in which a tamoxifen-inducible Cre-recombinase is expressed under the control of the Cx3cr1 promoter: Cx3cr1 CreER/+:R26iDT-A/+ and Cx3cr1 CreER/+:R26iDTR/+ . When activated by tamoxifen, nuclear translocation of the CreER fusion protein is transient and recombination occurs only for a limited period, so only long-lived cells as microglia, but not peripheral macrophages with a shorter lifespan, will be depleted. Later, the generation of conditional knockout mice harboring a loxP-flanked exon within the Csf1r gene ( Csf1r flx/flx ) has allowed spatial and temporal control of microglia upon combination with the appropriate Cre lines [51]. Additionally, the targeting of a more specific microglia-signature gene, as Tmem119 , has allowed the generation of Tmem119 CreERT2 lines [52]. These new genetic models considerably represent an improvement in the manipulation of microglia providing a valuable tool for the functional study of these cells (reviewed in [37][38]).
3. Microglial Depletion as a Model of Microglial Replacement
Due to high mouse microglial proliferation capacity, these depletion models do not mimic a situation of microglial degeneration, as previously desired, but a context of microglial renewal. The characterization of emerging microglia’s capabilities will allow us to validate the efficacy of microglial depletion-repopulation strategies as potential therapeutic tools. This approach will be beneficial when microglia are hyperactive as well as in a context of microglial degeneration [53] or senescent [54], because in both cases microglia may contribute to neuron toxicity. As microglial chronic activation is sustained by an oxidative metabolism [18], this may compromise oxygen availability for other cellular populations. Additionally, an excessive microgliosis may induce microglial mitochondrial damage and be a major source of reactive oxidative species [55], leading to oxidative damage in neurons, to astrocyte reactivity [56] and to an exacerbation of the inflammatory cascade.
Repopulating microglia appear to fulfil functions of resident microglia and is capable of monitoring the environment and responding to acute stimuli [57][58][59]. Adult newborn microglia have been described to gradually regain steady-state maturity, transcriptionally clustering close to control microglia 2 weeks after depletion [60]. Zhan et al. (2019) also showed that the restoration of microglial homeostatic density requires NF-κB signaling as well as apoptotic egress of excessive cells [60]. In accordance, Huang et al. (2018) found no transcriptomic differences in repopulated microglia (2-month after depletion treatment) compared to resident microglia, neither in resting conditions nor after LPS challenge [61]. More recently, Gratuze et al. (2021) also described a homeostatic gene signature and equal ability to cluster around amyloid deposits in repopulated microglia in an AD mouse model [62].
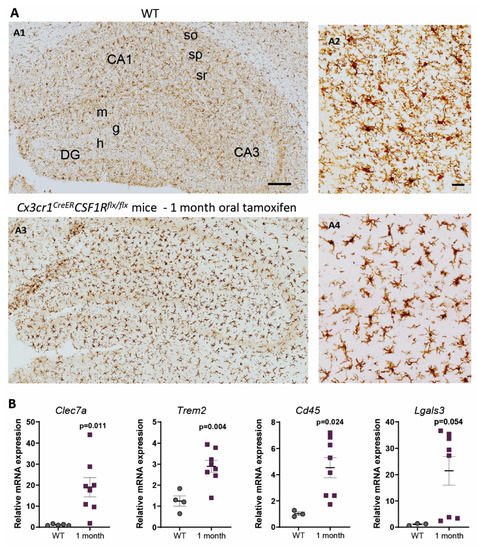
In contrast, under 1-month continuous tamoxifen treatment in our Cx3cr1 CreER /Csf1r flx/flx model, we observed that emerging microglia displayed an active morphology, with a thickening of cell body and shortening and thickening of microglial processes ( Figure2 A). Furthermore, the increased expression of Clec7a , Cd45 , Trem2 and Lgals3 corroborated microglial activation after 1-month treatment ( Figure 2 B). We should take into account that, unlike previously mentioned publications, we maintained the depletion inductor, so a continuous microglial depletion and repopulation was taking place. An active phenotype is typical of phagocytic microglia, which could be eliminating cellular debris of dead microglia [29]. In this sense, an upregulation of scavenger-associated proteins, such as Cd36 [61], and of the phagocytic marker Cd68 [46] have been reported in repopulating microglia. However, given the small remaining microglial population at some points (for instance, at the end of our acute treatment ( Figure 1 A,B)), the CNS must count with an additional mechanism to eliminate all cellular debris, among which phagocytic activity of astrocytes has been proposed [63][64]. Konishi et al. (2020) demonstrated that, after specific microglial depletion (using Siglech dtr mice), astrocytes, rather than CNS-associated macrophages or circulating monocytes, clear microglial debris [63].
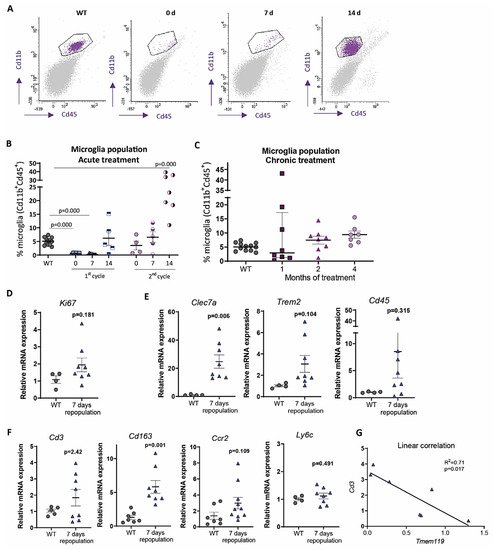
Finally, when drawing conclusion from chronic depletion models, we must be aware that changes observed are difficult to be unequivocally addressed to one factor. They may be due to: (i) a reduction in total microglial levels, (ii) a reduction of previous, sometimes burnt out, microglia, or (iii) the presence of emerging microglia whose characteristics are still not thoroughly described. Same challenges will occur with other state-of-the-art microglial manipulation techniques, such as chimeric mice. These models have been proposed as emerging tools to substitute exhausted microglia and/or to characterize human microglial response in AD pathology. Under this approach, mouse microglia is depleted to be replaced with human iPS-derived microglia in immunosuppressed mice [65]. After this manipulation, alterations in the progression of AD pathologies could be addressed to: (i) a reduction in the number of mouse microglia, (ii) the effect of human microglia, (iii) the immunosuppression of mice, (iv) the interaction between mouse and human microglia. Additionally, taking into account the stablished interaction between microglia and astrocytes [66], in depletion as well as in chimeric mice, modifications in the pathologies may also be caused by changes occurred in other glial cells. In essence, although they are promising tools, we should be cautious when drawing conclusions from these models.
4. Do Microglia Refresh or Poison AD Progression?
In order to further characterize microglial role in AD, microglial depletion models are being combined with AD mouse models bearing either Aβ and/or Tau pathologies ( Table 1 ). Currently, controversial data are found and new studies are needed to clarify if microglia is beneficial or detrimental to AD pathology.
| Pathology | Depletion Model | Outcomes | References |
|---|---|---|---|
| Aβ | PLX5622 in 5xFAD mice, from 4- to 5-month-old. | 50% microglia depletion. Reduction of microgliosis and plaque burden, enhancement of neuritic dystrophies. | [67] |
| Aβ | PLX3397 in 5xFAD mice, from 9- to 10-month-old. | Around 50% microglia depletion. Decrease in Aβ deposition and rescue of dopaminergic signaling. | [68] |
| Aβ | PLX5622 in APP/PS1 mice, from 12- to 13-month-old. | Diminution of leukotriene biosynthesis and the neuronal 5-lipoxygenase. | [69] |
| Aβ | PLX5622 in 5xFAD mice from 1.5 to 4- or 7-month-olds. | 97% microglia depletion. Reduction of plaque deposition, but increase of cerebral amyloid angiopathy formation. | [42] |
| Aβ | PLX3397 in 5xFAD mice from 2- to 5-month-old. | 70–80% microglia depletion. Reduction of intraneuronal amyloid, neuritic plaque deposition and improvement in cognitive functions (fear conditioning tests). | [70] |
| Aβ | Diphtheria toxin in 15 months-old Cx3cr1CreER/+:R26DTR/+/APPxPS1 mice, for 1–2 weeks. | 90% depletion. No changes in the number of Aβ plaques, but an increase in size. | [71] |
| Aβ | GWS2580 in APP/PS1 mice from 6- to 9-month-old. | 30% reduction of microglia. No changes in the number of Aβ plaques. Improved performance in memory and behavioral tasks. | [40] |
| Aβ | PLX3397 in 5xFAD from 10- to 11-month-old. | 90% microglia depletion. No alterations in β-amyloid levels or plaque load, but rescue of dendritic spine loss and improvements in contextual memory. | [72] |
| Aβ and Tau | PLX5562 in 3xTg mice for 3 months. | 30% microglia depletion. No changes in total or phosphorylated Tau. Improvements in cognition. | [73] |
| Aβ and Tau | PLX3397 from 5.5- to 7-month-old in 5xFAD/PS19 Tau -injected mice. | 81% microglia depletion. Higher reduction in non-plaque-associated microglia. No changes in Aβ pathology, reduction in Tau pathology and neurodegeneration. | [74] |
| Aβ and Tau | PLX3397 from 6- to 9-month-old in 5xFAD mice injected with AD-Tau. | Improved cognitive and neuronal deficits. Enhancement of Tau seeding and spreading around plaques. | [62] |
| Tau | Cx3cr1CreER/R26DTA/hTAU mice, treated with tamoxifen for 2–3 months at different ages. | 60% microglia depletion. No changes in soluble oligomeric, phosphorylated or total aggregated Tau levels. | [75] |
| Tau | PLX3397 in P301S APOE E4 mice from 6- to 9-month-old. | Total microglia depletion. Protection from brain volume loss and neurodegeneration. Reduction of Tau pathology progression. | [46] |
| Tau | PLX3397 in rTg4510 mice, from 12- to 15-month-old. | 30% microglia depletion. No changes in Tau burden, cortical atrophy, blood vessels or glial activation. | [76] |
| Tau | (a) Clodronate liposomes and PLX3397 in AAV-GFP/Tau injected C57BL/6 mice. (b) PLX3397 in PS19 mice. In both cases, from 3.5- to 4.5-month-old. | 70–80% (a) and 90% (b) microglia depletion. Reduction of phospho-Tau. | [77] |
Relevant results in AD mouse models of microglia depletion. Mouse models used and main outcomes are shown.
Studies of microglial depletion in other CNS pathologies showed similarly controversial results. Rice et al. (2017) and Acharya et al. (2016) described significant improvements subsequent to microglial depletion in neuronal damage models [78][79]. In the same line, Li et al. (2017) showed neuroprotection following microglial depletion in a model of intracerebral hemorrhage [80]. However, other publications reported an increase in neuroinflammation and brain damage after microglial depletion in ischemia models [81][82].
Therefore, novel approaches to increase microglial renewal may be a promising tool to assess microglial role in the ATN continuum and to design potential therapies for AD.
References
- Nguyen, A.T.; Wang, K.; Hu, G.; Wang, X.; Miao, Z.; Azevedo, J.A.; Suh, E.; Van Deerlin, V.M.; Choi, D.; Roeder, K.; et al. APOE and TREM2 Regulate Amyloid-Responsive Microglia in Alzheimer’s Disease. Acta Neuropathol. 2020.
- Wang, H.; Dey, K.K.; Chen, P.-C.; Li, Y.; Niu, M.; Cho, J.-H.; Wang, X.; Bai, B.; Jiao, Y.; Chepyala, S.R.; et al. Integrated Analysis of Ultra-Deep Proteomes in Cortex, Cerebrospinal Fluid and Serum Reveals a Mitochondrial Signature in Alzheimer’s Disease. Mol. Neurodegener. 2020, 15, 43.
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate Mapping Analysis Reveals That Adult Microglia Derive from Primitive Macrophages. Science 2010, 330, 841–845.
- Lawson, L.J.; Perry, V.H.; Dri, P.; Gordon, S. Heterogeneity in the Distribution and Morphology of Microglia in the Normal Adult Mouse Brain. Neuroscience 1990, 39, 151–170.
- Askew, K.; Li, K.; Olmos-Alonso, A.; Garcia-Moreno, F.; Liang, Y.; Richardson, P.; Tipton, T.; Chapman, M.A.; Riecken, K.; Beccari, S.; et al. Coupled Proliferation and Apoptosis Maintain the Rapid Turnover of Microglia in the Adult Brain. Cell Rep. 2017, 18, 391–405.
- Réu, P.; Khosravi, A.; Bernard, S.; Mold, J.E.; Salehpour, M.; Alkass, K.; Perl, S.; Tisdale, J.; Possnert, G.; Druid, H.; et al. The Lifespan and Turnover of Microglia in the Human Brain. Cell Rep. 2017, 20, 779–784.
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical Model of Dynamic Biomarkers of the Alzheimer’s Pathological Cascade. Lancet Neurol. 2010, 9, 119–128.
- He, Z.; Guo, J.L.; McBride, J.D.; Narasimhan, S.; Kim, H.; Changolkar, L.; Zhang, B.; Gathagan, R.J.; Yue, C.; Dengler, C.; et al. Amyloid-β Plaques Enhance Alzheimer’s Brain Tau-Seeded Pathologies by Facilitating Neuritic Plaque Tau Aggregation. Nat. Med. 2018, 24, 29–38.
- Clayton, K.; Delpech, J.C.; Herron, S.; Iwahara, N.; Ericsson, M.; Saito, T.; Saido, T.C.; Ikezu, S.; Ikezu, T. Plaque Associated Microglia Hyper-Secrete Extracellular Vesicles and Accelerate Tau Propagation in a Humanized APP Mouse Model. Mol. Neurodegener. 2021, 16, 18.
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-Cell Transcriptomic Analysis of Alzheimer’s Disease. Nature 2019, 570, 332–337.
- Grubman, A.; Chew, G.; Ouyang, J.F.; Sun, G.; Choo, X.Y.; McLean, C.; Simmons, R.K.; Buckberry, S.; Vargas-Landin, D.B.; Poppe, D.; et al. A Single-Cell Atlas of Entorhinal Cortex from Individuals with Alzheimer’s Disease Reveals Cell-Type-Specific Gene Expression Regulation. Nat. Neurosci. 2019, 22, 2087–2097.
- Masuda, T.; Sankowski, R.; Staszewski, O.; Böttcher, C.; Amann, L.; Sagar; Scheiwe, C.; Nessler, S.; Kunz, P.; van Loo, G.; et al. Spatial and Temporal Heterogeneity of Mouse and Human Microglia at Single-Cell Resolution. Nature 2019, 566, 388–392.
- Leng, F.; Edison, P. Neuroinflammation and Microglial Activation in Alzheimer Disease: Where Do We Go from Here? Nat. Rev. Neurol. 2021, 17, 157–172.
- Zhou, Y.; Song, W.M.; Andhey, P.S.; Swain, A.; Levy, T.; Miller, K.R.; Poliani, P.L.; Cominelli, M.; Grover, S.; Gilfillan, S.; et al. Human and Mouse Single-Nucleus Transcriptomics Reveal TREM2-Dependent and—Independent Cellular Responses in Alzheimer’s Disease. Nat. Med. 2020, 26, 131–142.
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17.
- Condello, C.; Yuan, P.; Schain, A.; Grutzendler, J. Microglia Constitute a Barrier That Prevents Neurotoxic Protofibrillar Aβ42 Hotspots around Plaques. Nat. Commun. 2015, 6, 6176.
- Deczkowska, A.; Keren-Shaul, H.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 2018, 173, 1073–1081.
- March-Diaz, R.; Lara-Ureña, N.; Romero-Molina, C.; Heras-Garvin, A.; Luis, C.O.S.; Alvarez-Vergara, M.I.; Sanchez-Garcia, M.A.; Sanchez-Mejias, E.; Davila, J.C.; Rosales-Nieves, A.E.; et al. Hypoxia Compromises the Mitochondrial Metabolism of Alzheimer’s Disease Microglia via HIF1. Nat. Aging 2021, 1, 385–399.
- Friedman, B.A.; Srinivasan, K.; Ayalon, G.; Meilandt, W.J.; Lin, H.; Huntley, M.A.; Cao, Y.; Lee, S.-H.; Haddick, P.C.G.; Ngu, H.; et al. Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Rep. 2018, 22, 832–847.
- Romero-Molina, C.; Navarro, V.; Sanchez-Varo, R.; Jimenez, S.; Fernandez-Valenzuela, J.J.; Sanchez-Mico, M.V.; Muñoz-Castro, C.; Gutierrez, A.; Vitorica, J.; Vizuete, M. Distinct Microglial Responses in Two Transgenic Murine Models of TAU Pathology. Front. Cell. Neurosci. 2018, 12, 421.
- Leyns, C.E.G.; Gratuze, M.; Narasimhan, S.; Jain, N.; Koscal, L.J.; Jiang, H.; Manis, M.; Colonna, M.; Lee, V.M.Y.; Ulrich, J.D.; et al. TREM2 Function Impedes Tau Seeding in Neuritic Plaques. Nat. Neurosci. 2019, 22, 1217–1222.
- Serrano-Pozo, A.; Gómez-Isla, T.; Growdon, J.H.; Frosch, M.P.; Hyman, B.T. A Phenotypic Change but Not Proliferation Underlies Glial Responses in Alzheimer Disease. Am. J. Pathol. 2013, 182, 2332–2344.
- Hamelin, L.; Lagarde, J.; Dorothée, G.; Leroy, C.; Labit, M.; Comley, R.A.; de Souza, L.C.; Corne, H.; Dauphinot, L.; Bertoux, M.; et al. Early and Protective Microglial Activation in Alzheimer’s Disease: A Prospective Study Using 18F-DPA-714 PET Imaging. Brain J. Neurol. 2016, 139, 1252–1264.
- Fan, Z.; Brooks, D.J.; Okello, A.; Edison, P. An Early and Late Peak in Microglial Activation in Alzheimer’s Disease Trajectory. Brain 2017, 140, 792–803.
- Navarro, V.; Sanchez-Mejias, E.; Jimenez, S.; Muñoz-Castro, C.; Sanchez-Varo, R.; Davila, J.C.; Vizuete, M.; Gutierrez, A.; Vitorica, J. Microglia in Alzheimer’s Disease: Activated, Dysfunctional or Degenerative. Front. Aging Neurosci. 2018, 10, 140.
- Jimenez, S.; Baglietto-Vargas, D.; Caballero, C.; Moreno-Gonzalez, I.; Torres, M.; Sanchez-Varo, R.; Ruano, D.; Vizuete, M.; Gutierrez, A.; Vitorica, J. Inflammatory Response in the Hippocampus of PS1M146L/APP751SL Mouse Model of Alzheimer’s Disease: Age-Dependent Switch in the Microglial Phenotype from Alternative to Classic. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 11650–11661.
- Galatro, T.F.; Holtman, I.R.; Lerario, A.M.; Vainchtein, I.D.; Brouwer, N.; Sola, P.R.; Veras, M.M.; Pereira, T.F.; Leite, R.E.P.; Möller, T.; et al. Transcriptomic Analysis of Purified Human Cortical Microglia Reveals Age-Associated Changes. Nat. Neurosci. 2017, 20, 1162–1171.
- Bohlen, C.J.; Bennett, F.C.; Tucker, A.F.; Collins, H.Y.; Mulinyawe, S.B.; Barres, B.A. Diverse Requirements for Microglial Survival, Specification, and Function Revealed by Defined-Medium Cultures. Neuron 2017, 94, 759–773.
- Elmore, M.R.P.; Najafi, A.R.; Koike, M.A.; Dagher, N.N.; Spangenberg, E.E.; Rice, R.A.; Kitazawa, M.; Matusow, B.; Nguyen, H.; West, B.L.; et al. CSF1 Receptor Signaling Is Necessary for Microglia Viability, Which Unmasks a Cell That Rapidly Repopulates the Microglia-Depleted Adult Brain. Neuron 2014, 82, 380–397.
- Wu, L.; Li, Y.; Yu, M.; Yang, F.; Tu, M.; Xu, H. Notch Signaling Regulates Microglial Activation and Inflammatory Reactions in a Rat Model of Temporal Lobe Epilepsy. Neurochem. Res. 2018, 43, 1269–1282.
- Elegheert, J.; Desfosses, A.; Shkumatov, A.V.; Wu, X.; Bracke, N.; Verstraete, K.; Van Craenenbroeck, K.; Brooks, B.R.; Svergun, D.I.; Vergauwen, B.; et al. Extracellular Complexes of the Hematopoietic Human and Mouse CSF-1 Receptor Are Driven by Common Assembly Principles. Structure 2011, 19, 1762–1772.
- Felix, J.; De Munck, S.; Verstraete, K.; Meuris, L.; Callewaert, N.; Elegheert, J.; Savvides, S.N. Structure and Assembly Mechanism of the Signaling Complex Mediated by Human CSF-1. Structure 2015, 23, 1621–1631.
- Ma, X.; Lin, W.Y.; Chen, Y.; Stawicki, S.; Mukhyala, K.; Wu, Y.; Martin, F.; Bazan, J.F.; Starovasnik, M.A. Structural Basis for the Dual Recognition of Helical Cytokines IL-34 and CSF-1 by CSF-1R. Structure 2012, 20, 676–687.
- Patel, S.; Player, M. Colony-Stimulating Factor-1 Receptor Inhibitors for the Treatment of Cancer and Inflammatory Disease. Curr. Top. Med. Chem. 2009, 9.
- Chitu, V.; Stanley, E.R. Chapter Seven—Regulation of Embryonic and Postnatal Development by the CSF-1 Receptor. In Current Topics in Developmental Biology; Jenny, A., Ed.; Protein Kinases in Development and Disease; Academic Press: Cambridge, MA, USA, 2017; Volume 123, pp. 229–275.
- Erblich, B.; Zhu, L.; Etgen, A.M.; Dobrenis, K.; Pollard, J.W. Absence of Colony Stimulation Factor-1 Receptor Results in Loss of Microglia, Disrupted Brain Development and Olfactory Deficits. PLoS ONE 2011, 6, e26317.
- Green, K.N.; Crapser, J.D.; Hohsfield, L.A. To Kill a Microglia: A Case for CSF1R Inhibitors. Trends Immunol. 2020, 41, 771–784.
- Wu, W.; Li, Y.; Wei, Y.; Bosco, D.B.; Xie, M.; Zhao, M.-G.; Richardson, J.R.; Wu, L.-J. Microglial Depletion Aggravates the Severity of Acute and Chronic Seizures in Mice. Brain. Behav. Immun. 2020, 89, 245–255.
- Han, J.; Zhu, K.; Zhang, X.-M.; Harris, R.A. Enforced Microglial Depletion and Repopulation as a Promising Strategy for the Treatment of Neurological Disorders. Glia 2019, 67, 217–231.
- Olmos-Alonso, A.; Schetters, S.T.T.; Sri, S.; Askew, K.; Mancuso, R.; Vargas-Caballero, M.; Holscher, C.; Perry, V.H.; Gomez-Nicola, D. Pharmacological Targeting of CSF1R Inhibits Microglial Proliferation and Prevents the Progression of Alzheimer’s-like Pathology. Brain J. Neurol. 2016, 139, 891–907.
- Mancuso, R.; Fryatt, G.; Cleal, M.; Obst, J.; Pipi, E.; Monzón-Sandoval, J.; Ribe, E.; Winchester, L.; Webber, C.; Nevado, A.; et al. CSF1R Inhibitor JNJ-40346527 Attenuates Microglial Proliferation and Neurodegeneration in P301S Mice. Brain 2019, 142, 3243–3264.
- Spangenberg, E.; Severson, P.L.; Hohsfield, L.A.; Crapser, J.; Zhang, J.; Burton, E.A.; Zhang, Y.; Spevak, W.; Lin, J.; Phan, N.Y.; et al. Sustained Microglial Depletion with CSF1R Inhibitor Impairs Parenchymal Plaque Development in an Alzheimer’s Disease Model. Nat. Commun. 2019, 10, 3758.
- Lei, F.; Cui, N.; Zhou, C.; Chodosh, J.; Vavvas, D.G.; Paschalis, E.I. CSF1R Inhibition by a Small-Molecule Inhibitor Is Not Microglia Specific; Affecting Hematopoiesis and the Function of Macrophages. Proc. Natl. Acad. Sci. USA 2020, 117, 23336–23338.
- Thompson, M.L.; Jimenez-Andrade, J.M.; Chartier, S.; Tsai, J.; Burton, E.A.; Habets, G.; Lin, P.S.; West, B.L.; Mantyh, P.W. Targeting Cells of the Myeloid Lineage Attenuates Pain and Disease Progression in a Prostate Model of Bone Cancer. Pain 2015, 156, 1692–1702.
- Chitu, V.; Gokhan, Ş.; Nandi, S.; Mehler, M.F.; Stanley, E.R. Emerging Roles for CSF-1 Receptor and Its Ligands in the Nervous System. Trends Neurosci. 2016, 39, 378–393.
- Shi, Y.; Manis, M.; Long, J.; Wang, K.; Sullivan, P.M.; Serrano, J.R.; Hoyle, R.; Holtzman, D.M. Microglia Drive APOE-Dependent Neurodegeneration in a Tauopathy Mouse Model. J. Exp. Med. 2019, 216, 2546–2561.
- Jäkel, S.; Dimou, L. Glial Cells and Their Function in the Adult Brain: A Journey through the History of Their Ablation. Front. Cell. Neurosci. 2017, 11, 24.
- Gowing, G.; Vallières, L.; Julien, J.-P. Mouse Model for Ablation of Proliferating Microgliain Acute CNS Injuries. Glia 2006, 53, 331–337.
- Bruttger, J.; Karram, K.; Wörtge, S.; Regen, T.; Marini, F.; Hoppmann, N.; Klein, M.; Blank, T.; Yona, S.; Wolf, Y.; et al. Genetic Cell Ablation Reveals Clusters of Local Self-Renewing Microglia in the Mammalian Central Nervous System. Immunity 2015, 43, 92–106.
- Kim, H.; Kim, M.; Im, S.-K.; Fang, S. Mouse Cre-LoxP System: General Principles to Determine Tissue-Specific Roles of Target Genes. Lab. Anim. Res. 2018, 34, 147–159.
- Cronk, J.C.; Filiano, A.J.; Louveau, A.; Marin, I.; Marsh, R.; Ji, E.; Goldman, D.H.; Smirnov, I.; Geraci, N.; Acton, S.; et al. Peripherally Derived Macrophages Can Engraft the Brain Independent of Irradiation and Maintain an Identity Distinct from Microglia. J. Exp. Med. 2018, 215, 1627–1647.
- Kaiser, T.; Feng, G. Tmem119-EGFP and Tmem119-CreERT2 Transgenic Mice for Labeling and Manipulating Microglia. eNeuro 2019, 6.
- Sanchez-Mejias, E.; Navarro, V.; Jimenez, S.; Sanchez-Mico, M.; Sanchez-Varo, R.; Nuñez-Diaz, C.; Trujillo-Estrada, L.; Davila, J.C.; Vizuete, M.; Gutierrez, A.; et al. Soluble Phospho-Tau from Alzheimer’s Disease Hippocampus Drives Microglial Degeneration. Acta Neuropathol. 2016, 132, 897–916.
- Hu, Y.; Fryatt, G.L.; Ghorbani, M.; Obst, J.; Menassa, D.A.; Martin-Estebane, M.; Muntslag, T.A.O.; Olmos-Alonso, A.; Guerrero-Carrasco, M.; Thomas, D.; et al. Replicative Senescence Dictates the Emergence of Disease-Associated Microglia and Contributes to Aβ Pathology. Cell Rep. 2021, 35.
- Ghosh, S.; Castillo, E.; Frias, E.S.; Swanson, R.A. Bioenergetic Regulation of Microglia. Glia 2018, 66, 1200–1212.
- Joshi, A.U.; Minhas, P.S.; Liddelow, S.A.; Haileselassie, B.; Andreasson, K.I.; Dorn, G.W.; Mochly-Rosen, D. Fragmented Mitochondria Released from Microglia Trigger A1 Astrocytic Response and Propagate Inflammatory Neurodegeneration. Nat. Neurosci. 2019, 22, 1635–1648.
- Elmore, M.R.P.; Lee, R.J.; West, B.L.; Green, K.N. Characterizing Newly Repopulated Microglia in the Adult Mouse: Impacts on Animal Behavior, Cell Morphology, and Neuroinflammation. PLoS ONE 2015, 10, e0122912.
- Varvel, N.H.; Grathwohl, S.A.; Baumann, F.; Liebig, C.; Bosch, A.; Brawek, B.; Thal, D.R.; Charo, I.F.; Heppner, F.L.; Aguzzi, A.; et al. Microglial Repopulation Model Reveals a Robust Homeostatic Process for Replacing CNS Myeloid Cells. Proc. Natl. Acad. Sci. USA 2012, 109, 18150–18155.
- Zhang, Y.; Zhao, L.; Wang, X.; Ma, W.; Lazere, A.; Qian, H.-H.; Zhang, J.; Abu-Asab, M.; Fariss, R.N.; Roger, J.E.; et al. Repopulating Retinal Microglia Restore Endogenous Organization and Function under CX3CL1-CX3CR1 Regulation. Sci. Adv. 2018, 4, eaap8492.
- Zhan, L.; Krabbe, G.; Du, F.; Jones, I.; Reichert, M.C.; Telpoukhovskaia, M.; Kodama, L.; Wang, C.; Cho, S.; Sayed, F.; et al. Proximal Recolonization by Self-Renewing Microglia Re-Establishes Microglial Homeostasis in the Adult Mouse Brain. PLoS Biol. 2019, 17, e3000134.
- Huang, Y.; Xu, Z.; Xiong, S.; Sun, F.; Qin, G.; Hu, G.; Wang, J.; Zhao, L.; Liang, Y.-X.; Wu, T.; et al. Repopulated Microglia Are Solely Derived from the Proliferation of Residual Microglia after Acute Depletion. Nat. Neurosci. 2018, 21, 530–540.
- Gratuze, M.; Chen, Y.; Parhizkar, S.; Jain, N.; Strickland, M.R.; Serrano, J.R.; Colonna, M.; Ulrich, J.D.; Holtzman, D.M. Activated Microglia Mitigate Aβ-Associated Tau Seeding and Spreading. J. Exp. Med. 2021, 218.
- Konishi, H.; Okamoto, T.; Hara, Y.; Komine, O.; Tamada, H.; Maeda, M.; Osako, F.; Kobayashi, M.; Nishiyama, A.; Kataoka, Y.; et al. Astrocytic Phagocytosis Is a Compensatory Mechanism for Microglial Dysfunction. EMBO J. 2020, 39, e104464.
- Sanchez-Mico, M.V.; Jimenez, S.; Gomez-Arboledas, A.; Muñoz-Castro, C.; Romero-Molina, C.; Navarro, V.; Sanchez-Mejias, E.; Nuñez-Diaz, C.; Sanchez-Varo, R.; Galea, E.; et al. Amyloid-β Impairs the Phagocytosis of Dystrophic Synapses by Astrocytes in Alzheimer’s Disease. Glia 2021, 69, 997–1011.
- Zhang, Y.; Cui, D. Evolving Models and Tools for Microglial Studies in the Central Nervous System. Neurosci. Bull. 2021.
- Vainchtein, I.D.; Molofsky, A.V. Astrocytes and Microglia: In Sickness and in Health. Trends Neurosci. 2020, 43, 144–154.
- Casali, B.T.; MacPherson, K.P.; Reed-Geaghan, E.G.; Landreth, G.E. Microglia Depletion Rapidly and Reversibly Alters Amyloid Pathology by Modification of Plaque Compaction and Morphologies. Neurobiol. Dis. 2020, 142, 104956.
- Son, Y.; Jeong, Y.J.; Shin, N.-R.; Oh, S.J.; Nam, K.R.; Choi, H.-D.; Choi, J.Y.; Lee, H.-J. Inhibition of Colony-Stimulating Factor 1 Receptor by PLX3397 Prevents Amyloid Beta Pathology and Rescues Dopaminergic Signaling in Aging 5xFAD Mice. Int. J. Mol. Sci. 2020, 21, 5553.
- Michael, J.; Unger, M.S.; Poupardin, R.; Schernthaner, P.; Mrowetz, H.; Attems, J.; Aigner, L. Microglia Depletion Diminishes Key Elements of the Leukotriene Pathway in the Brain of Alzheimer’s Disease Mice. Acta Neuropathol. Commun. 2020, 8, 129.
- Sosna, J.; Philipp, S.; Albay, R.; Reyes-Ruiz, J.M.; Baglietto-Vargas, D.; LaFerla, F.M.; Glabe, C.G. Early Long-Term Administration of the CSF1R Inhibitor PLX3397 Ablates Microglia and Reduces Accumulation of Intraneuronal Amyloid, Neuritic Plaque Deposition and Pre-Fibrillar Oligomers in 5XFAD Mouse Model of Alzheimer’s Disease. Mol. Neurodegener. 2018, 13, 11.
- Zhao, R.; Hu, W.; Tsai, J.; Li, W.; Gan, W.-B. Microglia Limit the Expansion of β-Amyloid Plaques in a Mouse Model of Alzheimer’s Disease. Mol. Neurodegener. 2017, 12, 47.
- Spangenberg, E.E.; Lee, R.J.; Najafi, A.R.; Rice, R.A.; Elmore, M.R.P.; Blurton-Jones, M.; West, B.L.; Green, K.N. Eliminating Microglia in Alzheimer’s Mice Prevents Neuronal Loss without Modulating Amyloid-β Pathology. Brain J. Neurol. 2016, 139, 1265–1281.
- Dagher, N.N.; Najafi, A.R.; Kayala, K.M.N.; Elmore, M.R.P.; White, T.E.; Medeiros, R.; West, B.L.; Green, K.N. Colony-Stimulating Factor 1 Receptor Inhibition Prevents Microglial Plaque Association and Improves Cognition in 3xTg-AD Mice. J. Neuroinflamm. 2015, 12, 139.
- Lodder, C.; Scheyltjens, I.; Stancu, I.C.; Botella Lucena, P.; Gutiérrez de Ravé, M.; Vanherle, S.; Vanmierlo, T.; Cremers, N.; Vanrusselt, H.; Brône, B.; et al. CSF1R Inhibition Rescues Tau Pathology and Neurodegeneration in an A/T/N Model with Combined AD Pathologies, While Preserving Plaque Associated Microglia. Acta Neuropathol. Commun. 2021, 9, 108.
- Zhu, K.; Pieber, M.; Han, J.; Blomgren, K.; Zhang, X.-M.; Harris, R.A.; Lund, H. Absence of Microglia or Presence of Peripherally-Derived Macrophages Does Not Affect Tau Pathology in Young or Old HTau Mice. Glia 2020, 68, 1466–1478.
- Bennett, R.E.; Bryant, A.; Hu, M.; Robbins, A.B.; Hopp, S.C.; Hyman, B.T. Partial Reduction of Microglia Does Not Affect Tau Pathology in Aged Mice. J. Neuroinflamm. 2018, 15, 311.
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kügler, S.; Ikezu, T. Depletion of Microglia and Inhibition of Exosome Synthesis Halt Tau Propagation. Nat. Neurosci. 2015, 18, 1584–1593.
- Rice, R.A.; Pham, J.; Lee, R.J.; Najafi, A.R.; West, B.L.; Green, K.N. Microglial Repopulation Resolves Inflammation and Promotes Brain Recovery after Injury. Glia 2017, 65, 931–944.
- Acharya, M.M.; Green, K.N.; Allen, B.D.; Najafi, A.R.; Syage, A.; Minasyan, H.; Le, M.T.; Kawashita, T.; Giedzinski, E.; Parihar, V.K.; et al. Elimination of Microglia Improves Cognitive Function Following Cranial Irradiation. Sci. Rep. 2016, 6, 31545.
- Li, M.; Li, Z.; Ren, H.; Jin, W.-N.; Wood, K.; Liu, Q.; Sheth, K.N.; Shi, F.-D. Colony Stimulating Factor 1 Receptor Inhibition Eliminates Microglia and Attenuates Brain Injury after Intracerebral Hemorrhage. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2017, 37, 2383–2395.
- Jin, W.-N.; Shi, S.X.-Y.; Li, Z.; Li, M.; Wood, K.; Gonzales, R.J.; Liu, Q. Depletion of Microglia Exacerbates Postischemic Inflammation and Brain Injury. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2017, 37, 2224–2236.
- Szalay, G.; Martinecz, B.; Lénárt, N.; Környei, Z.; Orsolits, B.; Judák, L.; Császár, E.; Fekete, R.; West, B.L.; Katona, G.; et al. Microglia Protect against Brain Injury and Their Selective Elimination Dysregulates Neuronal Network Activity after Stroke. Nat. Commun. 2016, 7, 11499.