Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 1 by Patrick Provost.
Using a modified RNA-sequencing (RNA-seq) approach, we discovered a new family of unusually short RNAs mapping to ribosomal RNA 5.8S, which we named dodecaRNAs (doRNAs), according to the number of core nucleotides (12 nt) their members contain.
- RNA sequencing
- small RNA
- non-coding RNA
- RT-qPCR
- 5.8S rRNA
1. Introduction
High-throughput sequencing (HTS) technologies revolutionized molecular biology and genetics by allowing the sequencing of entire genomes and transcriptomes [1,2,3,4,5,6][1][2][3][4][5][6]. The use of RNA sequencing (RNA-seq), in particular, markedly expanded the repertoire of non-coding RNA (ncRNA) species [7[7][8][9][10][11][12],8,9,10,11,12], which are now recognized as critical regulators of gene expression. ncRNAs have been reported, among other things, to control the binding of transcription factors and to regulate alternative splicing and messenger RNA (mRNA) translation [9[9][10][13][14][15],10,13,14,15], which allows cells to rapidly adjust their gene expression programming to respond and adapt to a changing environment, including cellular stress conditions [16]. Notably, ncRNAs are involved in cell proliferation and development, and thus, their impairment likely contributes to the etiology of various diseases, including cancer [17,18,19,20][17][18][19][20].
ncRNAs are classified according to their origin, length, and/or function [21,22,23,24][21][22][23][24]. Small RNAs (sRNAs) comprise transcripts less than 200 nucleotides (nt) in length. Small RNA sequences may be transcribed from dedicated sequences and promoters [25], or derived from several pre-existing RNA species, including mRNA introns or exons, transfer RNAs (tRNAs) or ribosomal RNAs (rRNAs) [26,27][26][27]. This yields an extremely diverse population of sRNAs most often involved in the specific recognition of nucleic acid targets through complementary base pairing [28,29][28][29].
Historically, the discovery of microRNAs (miRNAs, 19 to 24 nt) was delayed by half a century because of the dogma prevailing at the time stating that such short RNAs could not be biologically relevant, leading researchers to perceive them as mere degradation products [30,31][30][31]. Despite the now recognized importance of sRNAs and the lessons learned from the past, a comparable belief still exists today that arbitrarily draws the limit for function or interest to sRNAs longer than 16 nt. It is believed that any endogenous sequences shorter than 16 nt may not be specific, be mapped with confidence to the genome or have biological significance. Therefore, these are readily discarded either prior to library construction or from sequencing datasets in a systematic manner. This is done in order to improve the signal-to-noise ratio, improve the depth of sequencing or facilitate downstream computational analyses [32], allegedly without the risk of losing important information. These beliefs are strong and have tainted the standardized pipeline of most, if not all, HTS platforms and procedures currently available to researchers worldwide.
The very high abundance of rRNAs in biological samples—they may represent ~80% of total RNA of a cell—is also perceived with a negative a priori. Focusing on improving the sensitivity of HTS experiments, most researchers choose to eliminate rRNAs by using rRNA removal kits, which also eliminates the possibility of obtaining information on sRNAs derived from the most abundant cellular RNA. Known as small ribosomal RNAs (srRNAs), they form an emerging family of ncRNAs [27,33][27][33] that display essential functions in gene regulation and development [34,35][34][35]. Whether endogenous sRNA species shorter than 16 nt or derived from rRNA exist remains unknown.
2. sRNA-Seq Analyses Revealed the Existence of 12-nt and 13-nt sRNAs
In a previous study, we have used sRNA-seq analysis, and we investigated the sRNA profile (8–30 nt) of six different species (H. sapiens, M. musculus, D. melanogaster, A. thaliana, S. pombe, S. cerevisiae) from 11 samples, and revealed the existence of very small RNAs of discrete sizes, with a large part coming from rRNA (Lambert et al., manuscript submitted).
Together, in these RNA-seq data (Figure 1A), a relatively high abundance of 12-nt and 13-nt RNAs was observed in human, mouse, Drosophila and S. pombe samples, but not in A. thaliana and S. cerevisiae. Notably, 13-nt RNAs were more abundant than 12-nt RNAs in human and S. pombe samples, compared to mouse samples, where they seem to be equally represented. On the contrary, 12-nt RNAs were more abundant than 13-nt RNAs in Drosophila (Figure 1A,B). In fact, in these four organisms, these two-size ranges of RNAs (12 and 13 nt) represented between 22 to 74% of all RNAs sequenced in the 8 to 30 nt window of RNA length (Figure 1B); and the mouse neuronal N2a cell line is the most enriched in 12-nt and 13-nt RNAs, when added together.
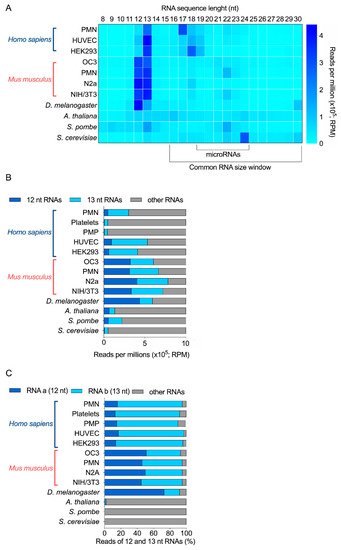
Figure 1. Relative abundance of 12 and 13 nt sRNA sequences obtained by sRNA-seq analyses of 11 different biological samples derived from 6 different species. (A) RPM abundance of RNA of 8 to 30 nt from 11 samples. (B) Relative abundance of 12-nt, 13-nt and other RNAs, expressed as RPM. (C) Relative proportion of the most abundant 12-nt RNA (RNA a) and 13-nt RNA (RNA b), compared with the other 12-nt and 13-nt RNAs detected by sRNA-seq (% of total reads). PMN, polymorphonuclear leukocytes; PMP, platelet-derived microparticles; HUVEC, human umbilical vein endothelial cells; HEK293, human embryonic kidney 293 cells; OC3, Old Cerebellum 3; N2a, mouse neuroblastoma cells; NIH/3T3, mouse embryonic fibroblast cells.
Further bioinformatics analyses revealed that a unique 12-nt RNA sequence represented at least 70% of all 12-nt long RNAs for H. sapiens, M. musculus and D. melanogaster. We observed the same results for 13-nt RNAs (Supplementary Figure S1). This abundant 13-nt sequence was the same as the 12-nt RNA, but with an extra Cytosine (C) at its 5′ end. The sequence of the human and mouse 12-nt and 13-nt RNAs was identical. In total, two sequences accounted for 90% RNA reads (Figure 1C). On the opposite, while 12-nt and 13-nt RNAs are detected in S. pombe, no specific sequence was more abundant than the others.
Our sRNA-seq data support the existence of very small RNAs that are 12 nt or 13 nt long, more abundant than microRNAs, and with species and cell specificity. In particular, two main sequences represented most of these 12-nt and 13-nt RNAs in human, mouse and Drosophila samples, but were absent from A. thaliana, S. pombe and S. cerevisiae.
3. The Two Most Abundant 12-nt and 13-nt Sequences Likely Derive from 5.8S Ribosomal RNA
First, screening of these 12-nt and 13-nt RNA sequences for vector, adaptor, linker and primer contamination did not yield any positive match, excluding the possibility that they represent an artifact (on genomic and transcript database). Using NCBI BLAST, we mapped the two most abundant 12-nt and 13-nt RNA sequences to the transcriptome of each organism. In human, both sequences perfectly matched with ribosomal RNAs; the 5.8S rRNA and its longer 45S rRNA precursor. The same results were obtained when mapping these sequences to the murine transcriptome, as both are conserved between the two species. In Drosophila, despite the difference in nucleotide composition, the corresponding, equivalently abundant 12-nt and 13-nt RNA sequences also matched to Drosophila’s 5.8S and 45S rRNAs (Table 1). The orthologous 12-nt Drosophila RNA differed from the corresponding human and mouse sequences by 2 nt, whereas the 13-nt Drosophila RNA harbor an Adenine (A) at its 5′ end, instead of a Cytosine (C) for the human and mouse—in both cases, the extra 5′ nucleotide matched to the corresponding nucleotide on the longer 45S rRNA (Figure 2).
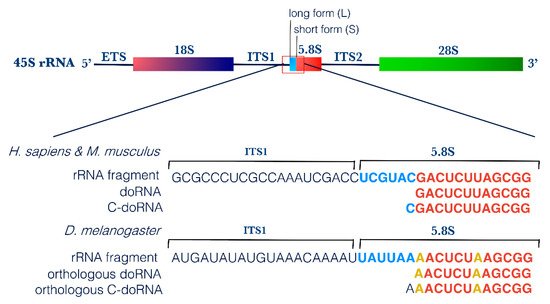
Figure 2. doRNA and C-doRNA sequences map to the 5′ end of the 5.8S rRNA. Schematic representation of doRNA and C-doRNA sequence alignment on the 45S rRNA in humans, mice and flies, using NCBI Nucleotide Reference Sequence (RefSeq) database. ETS, external transcribed spacer; ITS, internal transcribed spacer; rRNA, ribosomal RNA.
Table 1. Mapping of the doRNA and C-doRNA sequences to the human, mouse and fly transcriptomes using the nucleotide NCBI Basic Local Alignment Search Tool (BLAST) on NCBI database. The parameters used were the N blast, the standard database “nucleotide collection (nr/nt)” and the species “Homo sapiens,” or “Mus musculus,” or “Drosophila melanogaster.” The program used was BLASTN 2.12.0+. Results with 100% of identity and query coverage are shown in the table.
| Description | % Identity | Accession No. |
|---|
| Homo sapiens | ||
| Homo sapiens RNA, 5.8S ribosomal N3 (RNA5-8SN3), rRNA | 100% | NR_146153.1 |
| Homo sapiens RNA, 45S pre-ribosomal N2 (RNA45SN2), rRNA | 100% | NR_146144.1 |
| Mus musculus | ||
| Mus musculus 5.8S rRNA | 100% | K01367.1 |
| Mus musculus 18S rRNA, 5.8S rRNA and 28S rRNA | 100% | AH002077.2 |
| Drosophila melanogaster | ||
| Drosophila melanogaster pre-rRNA (pre-rRNA:CR45847), preRNA | 100% | NR_133558.1 |
| Drosophila melanogaster pre-rRNA (pre-rRNA:CR45846), preRNA | 100% | NR_133554.1 |
| Drosophila melanogaster pre-rRNA (pre-rRNA:CR45845), preRNA | 100% | NR_133549.1 |
| Drosophila melanogaster 5.8S rRNA (5.8SrRNA:CR45852) | 100% | NR_133551.1 |
| Drosophila melanogaster 5.8S and 2S rRNA | 100% | U20145.1 |
Pre-rRNA, rRNA precursor; rRNA, ribosomal RNA.
We propose to name this new RNA family dodecaRNAs (doRNAs), with respect to the number of core nucleotides (12 nt) their members contain. The most abundant 13-nt variant of doRNA harbors, in human and mouse, a C at its 5′ end and was consequently termed C-doRNA. We found that the doRNA sequence mapped directly to the 5′ end of the 5.8S rRNA (Figure 2). Thus, doRNAs might be formed through a specific and controlled cleavage of the 5.8S rRNA or transient rRNA precursors leading to 5.8S rRNA (e.g., 45S, 36S, 32S, 12S, 8S rRNAs). As rRNAs are the most abundant RNAs in cells (80% of all RNAs) [38][36], it is very likely that the doRNA and C-doRNA sequences, which are similarly overly abundant (e.g., compared to microRNAs) in the 8- to 30-nt window of RNA sizes, are sRNA fragments derived from rRNA (rRFs) [27].
This possibility is reinforced by the presence, in human and mouse 5.8S rRNA sequences, of a recurrent 2′-O-ribose methylation of the Uracil (U) positioned immediately downstream to the 3′ nucleotide of doRNAs [39][37]. This feature suggests that the modified U may be a signal or a determinant for the generation of their 3′ extremity (Figure 2). This feature reinforces the possibility that doRNAs originate from the 5.8S rRNA or longer precursors containing it.
References
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351.
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921.
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108.
- Waterston, R.H.; Lindblad-Toh, K.; Birney, E.; Rogers, J.; Abril, J.F.; Agarwal, P.; Agarwala, R.; Ainscough, R.; Alexandersson, M.; An, P.; et al. Initial sequencing and comparative analysis of the mouse genome. Nature 2002, 420, 520–562.
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; et al. The Transcriptional Landscape of the Mammalian Genome. Science 2005, 309, 1559–1563.
- Consortium, T.E.P.; Dunham, I.; Kundaje, A.; Aldred, S.F.; Collins, P.J.; Davis, C.A.; Doyle, F.; Epstein, C.B.; Frietze, S.; Harrow, J.; et al. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57.
- Zheng, Y. Introduction. In Computational Non-Coding RNA Biology; Zheng, Y., Ed.; Academic Press: Cambridge, MA, USA, 2019; p. 33. ISBN 978-0-12-814365-0.
- Wang, J.; Samuels, D.C.; Zhao, S.; Xiang, Y.; Zhao, Y.-Y.; Guo, Y. Current Research on Non-Coding Ribonucleic Acid (RNA). Genes 2017, 8, 366.
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789.
- Mattick, J.S. Non-coding RNAs: The architects of eukaryotic complexity. EMBO Rep. 2001, 2, 986–991.
- Delihas, N. Discovery and characterization of the first non-coding RNA that regulates gene expression, micF RNA: A historical perspective. World J. Biol. Chem. 2015, 6, 272–280.
- Zieve, G.; Penman, S. Small RNA species of the HeLa cell: Metabolism and subcellular localization. Cell 1976, 8, 19–31.
- Mattick, J.S. Challenging the dogma: The hidden layer of non-protein-coding RNAs in complex organisms. Bioessays 2003, 25, 930–939.
- Churko, J.M.; Mantalas, G.L.; Snyder, M.P.; Wu, J.C. Overview of high throughput sequencing technologies to elucidate molecular pathways in cardiovascular diseases. Circ. Res. 2013, 112, 1613–1623.
- Uchida, Y.; Chiba, T.; Kurimoto, R.; Asahara, H. Post-transcriptional regulation of inflammation by RNA-binding proteins via cis-elements of mRNAs. J. Biochem. 2019, 166, 375–382.
- Pirogov, S.A.; Gvozdev, V.A.; Klenov, M.S. Long Noncoding RNAs and Stress Response in the Nucleolus. Cells 2019, 8, 668.
- Veneziano, D.; Nigita, G.; Ferro, A. Computational Approaches for the Analysis of ncRNA through Deep Sequencing Techniques. Front. Bioeng. Biotechnol. 2015, 3, 77.
- Peng, J.-F.; Zhuang, Y.-Y.; Huang, F.-T.; Zhang, S.-N. Noncoding RNAs and pancreatic cancer. World J. Gastroenterol. 2016, 22, 801–814.
- Xie, Y.; Dang, W.; Zhang, S.; Yue, W.; Yang, L.; Zhai, X.; Yan, Q.; Lu, J. The role of exosomal noncoding RNAs in cancer. Mol. Cancer 2019, 18, 37.
- Tam, C.; Wong, J.H.; Tsui, S.K.W.; Zuo, T.; Chan, T.F.; Ng, T.B. LncRNAs with miRNAs in regulation of gastric, liver, and colorectal cancers: Updates in recent years. Appl. Microbiol. Biotechnol. 2019, 103, 4649–4677.
- St Laurent, G.; Wahlestedt, C.; Kapranov, P. The Landscape of long noncoding RNA classification. Trends Genet. 2015, 31, 239–251.
- Romano, G.; Veneziano, D.; Nigita, G.; Nana-Sinkam, S.P. RNA Methylation in ncRNA: Classes, Detection, and Molecular Associations. Front. Genet. 2018, 9, 243.
- Srijyothi, L.; Saravanaraman, P.; Prathama, T.; Cheemala, A.; Baluchamy, S. Roles of Non-Coding RNAs in Transcriptional Regulation; IntechOpen: London, UK, 2018; ISBN 978-1-78923-791-7.
- Liu, D.; Mewalal, R.; Hu, R.; Tuskan, G.A.; Yang, X. New technologies accelerate the exploration of non-coding RNAs in horticultural plants. Hortic. Res. 2017, 4, 17031.
- Ma, H.; Wu, Y.; Dang, Y.; Choi, J.-G.; Zhang, J.; Wu, H. Pol III Promoters to Express Small RNAs: Delineation of Transcription Initiation. Mol. Ther. Nucleic Acids 2014, 3, e161.
- Vickers, K.C.; Roteta, L.A.; Hucheson-Dilks, H.; Han, L.; Guo, Y. Mining diverse small RNA species in the deep transcriptome. Trends Biochem. Sci. 2015, 40, 4–7.
- Lambert, M.; Benmoussa, A.; Provost, P. Small Non-Coding RNAs Derived from Eukaryotic Ribosomal RNA. Non-Coding RNA 2019, 5, 16.
- Erdmann, V.A.; Barciszewska, M.Z.; Hochberg, A.; de Groot, N.; Barciszewski, J. Regulatory RNAs. Cell. Mol. Life Sci. 2001, 58, 960–977.
- Yamamura, S.; Imai-Sumida, M.; Tanaka, Y.; Dahiya, R. Interaction and cross-talk between non-coding RNAs. Cell. Mol. Life Sci. 2018, 75, 467–484.
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The, C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854.
- Reinhart, B.J.; Slack, F.J.; Basson, M.; Pasquinelli, A.E.; Bettinger, J.C.; Rougvie, A.E.; Horvitz, H.R.; Ruvkun, G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 2000, 403, 901–906.
- Dard-Dascot, C.; Naquin, D.; d’Aubenton-Carafa, Y.; Alix, K.; Thermes, C.; van Dijk, E. Systematic comparison of small RNA library preparation protocols for next-generation sequencing. BMC Genom. 2018, 19, 118.
- Pircher, A.; Gebetsberger, J.; Polacek, N. Ribosome-associated ncRNAs: An emerging class of translation regulators. RNA Biol. 2014, 11, 1335–1339.
- Locati, M.D.; Pagano, J.F.B.; Abdullah, F.; Ensink, W.A.; van Olst, M.; van Leeuwen, S.; Nehrdich, U.; Spaink, H.P.; Rauwerda, H.; Jonker, M.J.; et al. Identifying small RNAs derived from maternal- and somatic-type rRNAs in zebrafish development. Genome 2018, 61, 371–378.
- Li, S. Human 28s rRNA 5 terminal derived small RNA inhibits ribosomal protein mRNA levels. bioRxiv 2019.
- Zhao, S.; Zhang, Y.; Gamini, R.; Zhang, B.; von Schack, D. Evaluation of two main RNA-seq approaches for gene quantification in clinical RNA sequencing: PolyA+ selection versus rRNA depletion. Sci. Rep. 2018, 8, 4781.
- Nazar, R.N. The ribosomal 5.8S RNA: Eukaryotic adaptation or processing variant? Can. J. Biochem. Cell Biol. 1984, 62, 311–320.
More