Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Shelley Miyamoto and Version 2 by Lindsay Dong.
Cardiotoxicity is a well-recognized late effect among childhood cancer survivors. With various pediatric cancers becoming increasingly curable, it is imperative to understand the disease burdens that survivors may face in the future.
- cardio-oncology
- pediatric cancer
- cardiotoxicity
- anthracyclines
- pediatric heart failure
1. Introduction
Heart failure can be an early or late cardiotoxic side effect of cancer treatment in children. Anthracycline cardiotoxicity is characterized as acute if it occurs within the first week of treatment, early-onset progressive cardiotoxicity if it occurs within the first year of treatment completion, and late-onset progressive cardiotoxicity if it occurs more than one year after the completion of treatment [1].
2. Pathophysiology of Cardiotoxic Cancer Therapies
2.1. Anthracyclines
Among antineoplastic agents, anthracyclines are best-known for contributing to cardiotoxicity in childhood cancer survivors. This family of agents includes doxorubicin, daunorubicin, epirubicin, and idarubicin. In a landmark study in adults, Swain et al. reported an estimated cumulative percentage of doxorubicin-related congestive heart failure of 5% of patients who received a cumulative dose of 400 mg/m2 of doxorubicin, 26% of patients who received 550 mg/m2 of doxorubicin, and 48% of patients who received 700 mg/m2 of doxorubicin [2][10]. Reports from the Childhood Cancer Survivorship study have demonstrated a clear risk above cumulative doses of 250 mg/m2 but suggest that there may be no safe dose [3][5]. While there are many purported hypotheses of anthracyclines’ mechanisms of toxicity, a complete understanding of anthracycline-induced cardiomyopathy does not yet exist. Some of the more common implicated mechanisms will be briefly summarized here.
Anthracyclines increase cellular oxidative stress, which is associated with an imbalance between the creation of reactive oxygen species (ROS) and scavenging enzymes or antioxidants that normally keep ROS in check (Figure 1). Elevated ROS leads to oxidative stress as electrons are scavenged to reach a more stable state, leading to DNA damage, lipid membrane peroxidation, cell cycle arrest, telomere attrition, and apoptosis [4][11]. The most commonly used anthracycline in pediatric oncology is doxorubicin, which exerts its anti-neoplastic effect via topoisomerase IIα binding to DNA, inducing cell death. While tumor cells are rich in topoisomerase IIα, cardiomyocytes exhibit the topoisomerase IIβ isoform, which is thought to regulate genes involved in mitochondrial biogenesis and function [5][12]. Under doxorubicin’s influence, oxidative phosphorylation within the mitochondria is impacted and leads to an imbalance in favor of increased ROS, ultimately leading to cardiotoxicity [6][13]. Mitochondrial dysfunction also results from anthracyclines forming complexes with the inner mitochondrial membrane phospholipid cardiolipin, thereby disrupting electron transport chain activity and resulting in a shift to increased reliance on glucose metabolism in cardiomyocytes, which is a final common pathway in heart failure [7][8][14,15].
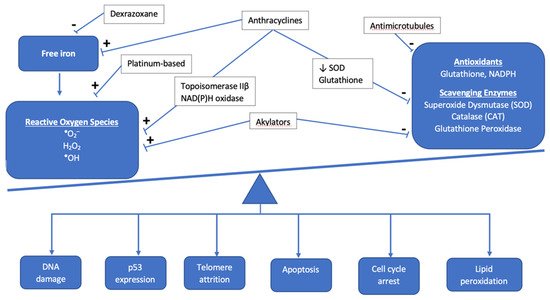
Figure 1. Overview of the interaction between antineoplastic chemotherapy and the role of oxidative stress in development of cardiotoxicity. The cumulative effect of chemotherapeutic agents results in the accumulation of reactive oxygen species and other mediators of oxidative stress, such as free iron, as antioxidants and scavenging enzymes are prevented from maintaining proper balance as they normally do under healthy physiologic conditions.
In addition to oxidative stress, there are other mechanisms of cellular apoptosis and cell injury mediated by anthracyclines. Anthracyclines cause direct DNA damage through the cleavage of DNA strands, but they also inhibit DNA biosynthesis and the enzymes involved in DNA repair [9][16]. Anthracyclines increase the likelihood that calcium channels in the sarcoplasmic reticulum are in the open state, resulting in an increase in calcium release [10][11][17,18]. Sustained calcium leak leads to further ROS production and ultimately culminates in the activation of the caspase cascade and cellular apoptosis. Anthracyclines result in impaired iron sequestration and, therefore, an increase in free iron accumulation in cardiomyocytes and mitochondria, further contributing to free radical generation, cell damage, and mitochondrial dysfunction [12][19]. Anthracyclines also impair nitric oxide and endothelin-1 production, contributing to endothelial cell dysfunction and diminished cardiomyocyte survival [13][20].
Genome wide association studies (GWAS) have contributed to the identification of possible genetic modifiers of risk for the development of anthracycline-induced cardiomyopathy; they are summarized in Table 1. In a recent review, Magdy and colleagues summarized the single nucleotide polymorphisms (SNPs) that increase the risk of cardiotoxicity [14][21]. For example, at the enzymatic level, the S427L variant of the RARG gene increases topoisomerase IIβ expression and is associated with worse cardiac outcomes among pediatric cancer survivors [15][22]. Variants in NAD(P)H oxidase subunits have been proposed to result in both acute and chronic forms of cardiotoxicity via ROS formation; [16][17][18][23,24,25] Cascales et al. examined cardiac histology among recently deceased patients treated with anthracyclines and discovered a five-fold increased odds (95% CI: 1.59–16.43) of cardiac interstitial fibrosis in the presence of the rs1883112 SNP in the p40phox subunit of NAD(P)H oxidase [19][26]. However, the rs4673 SNP in the p22phos subunit of NAD(P)H showed mixed results. While Cascales and colleagues noted a protective effect for those with the rs4673 SNP against myocardial fibrosis in the decedent samples (OR 0.11, 95% CI: 0.2–0.63), Wojnowski et al. found an association of this SNP with acute cardiotoxicity [16][19][23,26]. Moving from the enzymatic to the sarcomeric level, variants in the titin truncating gene (TTN) have been found to occur at higher rates in patients with cardiotoxicity than in the general population [20][27]. Garcia-Pavia subsequently confirmed the development of cardiomyopathy in TTN variant mice treated with anthracyclines [20][27].
Table 1. Overview of genetic modifiers of cardiotoxicity risk.
| Genetic Modifiers of Cardiotoxicity Risk | |
|---|---|
| Deleterious Effect | |
| Gene/SNP | Mechanism of toxicity |
| RARG (S427L variant) | Increase topoisomerase IIβ expression |
| rs1883112 SNP, p40phox subunit NAD(P)H oxidase | Interstitial fibrosis |
| UGT1A6*A | Decreased drug glucuronidation and clearance |
| MYH7 | Variants documented in familial dilated cardiomyopathy Downregulated in hiPSC-CM treated with doxorubicin |
| Titin truncating variants (TTN) | Encode A and I bands in sarcomeres; associated with depressed LV function |
| Indeterminant Effect | |
| rs4673 SNP, p22phos subunit NAD(P)H | Protective against myocardial fibrosis Acute cardiotoxicity |
Cardiotoxicity is also thought to arise from alterations in anthracycline transport and metabolism. In Magdy et al.’s review of the literature, they approximated that 45% of SNPs implicated in cardiotoxicity are associated with drug transport, which largely take the form of regulating the concentration of doxorubicin and its metabolites intracellularly [14][21].
2.2. Non-Anthracycline Agents
Many non-anthracycline chemotherapy agents are thought to exert their cardiotoxic effects via oxidative stress as well, though at much lower rates than their anthracycline counterparts. For example, a recent review, Zhang and colleagues summarize studies that suggest cyclophosphamide and its metabolites have the potential to both increase ROS production and interfere with the antioxidant system [21][32]. Cisplatin has been found to cause an accumulation of ROS, while anti-microtubule agents like vinblastine have been shown to decrease the activity of ROS scavenging enzymes [21][22][32,33].
Endothelial damage is thought to be a mechanism of cardiotoxicity mediated by non-anthracyclines as well. There is evidence that alkylators like cyclophosphamide directly damage endothelial cells, allowing for the extravasation of toxic metabolites and direct damage to myocytes [23][34]. While cardiotoxicity from 5-fluorouracil (5FU) is rare, it can occur and take the form of acute coronary syndrome, which may be mediated by nitric oxide synthase (NOS) causing coronary artery spasm, endothelium-dependent vasoconstriction, and ultimately, ischemia [24][25][35,36]. Finally, thromboembolism formation may be promoted by agents like cisplatin and cyclophosphamide, similarly predisposing to ischemia [23][34].
2.3. Radiation-Induced Cardiotoxicity
2.3.1. Mechanisms for Toxicity
Radiation therapy (RT) has improved oncologic outcomes for many pediatric cancers; however, RT to the chest is associated with a risk of cardiotoxicity. Radiation induces DNA damage, oxidative stress, endothelial cell senescence, and pro-inflammatory pathways. These changes may result in intimal thickening, fibrin deposition, lipid accumulation, and thrombosis [26][37]. In the acute time period, pericarditis and myocarditis may be observed; however, their incidence is low with modern RT. Late effects, such as coronary artery disease (CAD), valvular disease, restrictive cardiomyopathy, and arrhythmias may become apparent years or decades after RT [26][27][37,38]. Importantly, the age at onset of late effects is variable and depends in part on the patient’s age when cancer therapy commenced. While the median age of onset of cardiac disease in those exposed to radiation and other cardiotoxic therapies is generally in young adulthood, myocardial infarctions, congestive heart failure, pericardial disease, and valvular abnormalities can occur in the childhood years [3][5].
Other treatment and patient-specific factors increase the risk of radiation-associated cardiotoxicity. For example, the incidence of cardiotoxicity is higher in patients who received both anthracycline-based chemotherapy and RT compared to RT alone [28][29][39,40]. In addition, conventional risk factors, such as dyslipidemia, hypertension, and smoking, are independently associated with late cardiotoxicity [30][31][32][33][41,42,43,44]. Therefore, guidelines recommend regular screening for modifiable cardiac risk factors and the initiation of appropriate interventions in survivors who received RT to the chest [34][45].
2.3.2. Dose-Toxicity Relationship
Radiation-specific factors that influence the risk of cardiotoxicity include the total radiation dose to the heart, the volume of the heart exposed, and the dose per fraction [29][30][31][32][33][35][36][40,41,42,43,44,46,47] (Table 2). Recent data from the Childhood Cancer Survivor Study demonstrated a significant association between mean heart dose (MHD) and multiple adverse cardiac outcomes (heart failure, CAD, valvular disease, arrhythmia) in multivariable models that accounted for sex, treatment decade, anthracycline dose, and relevant comorbidities [30][41]. Importantly, this same study demonstrated that both the MHD and the risk of CAD in long-term survivors declined significantly over the study period. The decreased incidence of CAD with treatment decade was attenuated by adjustment for cardiac radiation exposure, suggesting that more modern RT is associated with a lower risk of CAD than historic RT [30][41].
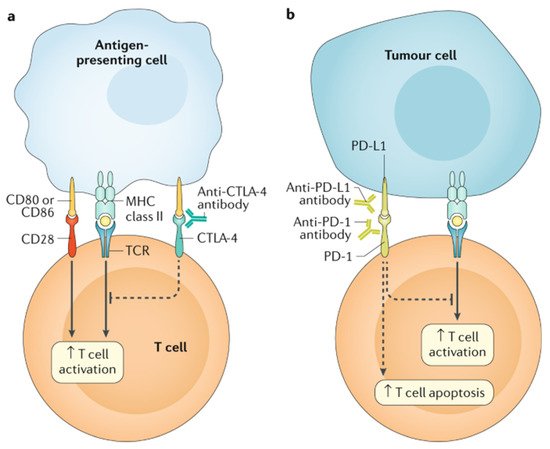
Table 2. Studies exploring the association of radiation dose with risk of grade ≥ 3 cardiac toxicity in adult survivors of childhood cancers. The reference group comprised adult survivors or childhood cancers without the cardiotoxic exposure. Higher radiation dose to the heart was associated with an increased risk of grade ≥ 3 cardiotoxicity. The risk was higher in patients who received anthracycline chemotherapy in addition to radiation therapy.
2.4. Targeted Cancer Therapies
| First Author | n | Treatment Period | Age at Treatment, Years | Length of Follow-Up, Years (Median) | Risk of Grade ≥ 3 Cardiotoxicity (95% CI) |
|---|---|---|---|---|---|
| Haddy [37] | 3162 | 1942–1985 | <17 | 26 | No anthracycline:
|
| van der Pal [38] | 1362 | 1966–1996 | <18 | 22 | Radiation only: HR 13.0 (2.8–61) Anthracycline & radiation: HR 49.5 (10.7–230) HR 1.8 (1.4–2.2) per 10 Gy to heart |
| Mulrooney [30] | 23,462 | 1970–1999 | <20 (median 6) | 28 | HF:
|
CAD: coronary artery disease; HF: heart failure; HR: hazard ratio; RR: relative risk.
2.4. Targeted Cancer Therapies
Targeted chemotherapy is more widely used in pediatric cancer, as our understanding of specific mutations in cancer development and the immune regulation to keep tumors in check is advanced. These therapies include immune checkpoint inhibitors, tyrosine kinase inhibitors (TKIs), and proteasome inhibitors. While there is great optimism for their role in revolutionizing cancer therapy, they are not without side effects, including cardiotoxicity. Immune checkpoint inhibitors work by blocking tumors’ attempts at evading the immune system, primarily by re-engaging T cell detection of tumors (this concept is further demonstrated in Figure 2). Tumors learn to express cytotoxic T-lymphocyte antigen 4 (CTLA-4), programmed cell death 1 (PD-1), and the ligand for PD-1 (PD-L1), all of which downregulate the immune response. Monoclonal antibodies against these receptors antagonize these downregulatory functions (effectively turning off the off switch) and allow the immune system to join in the fight against malignancy [39][50]. Examples include ipilimumab against CTLA-4 and nivolumab and pembrolizumab against PD-1.Figure 2. The main immunotherapy approaches that are approved for clinical use in cancer are the immune checkpoint inhibitors. These therapies are monoclonal antibodies that target the CTLA-4 and PD-1 receptors and the PD-1 ligand PD-L1, which are involved in the regulation of T cell activation. (a) T cell activation requires two signals: first, antigen recognition by the T cell receptor (TCR) following antigen presentation by major histocompatibility complex (MHC) class II molecules on the surface of antigen-presenting cells; and, second, signal modulation by CD80 or CD86 binding to the CD28 receptor. CTLA-4 is located on the T cell surface and competes with the CD28 receptor to bind CD80 or CD86, thereby blocking T cell activation. CTLA-4 inhibitors block CTLA-4–CD80 or CTLA-4–CD86 binding to facilitate T cell activation (dashed line). (b) PD-1 is a surface receptor that is expressed by T cells and promotes apoptosis of antigen-specific T cells and reduces apoptosis of regulatory T cells through its interaction with its ligand, PD-L1, which is expressed by tumour cells and myeloid cells. This interaction is useful in preventing autoimmunity in physiological conditions, but cancer cells exploit this process to escape from immune system activity upregulating PD-L1 expression. PD-1 and PD-L1 inhibitors block the PD-1–PD-L1 interaction, facilitating T cell activation and survival (dashed lines). (Figure with permission from Ramos-Casals M, Brahmer JR, et al. Immune-related adverse events of checkpoint inhibitors. Nat Rev Dis Primers. 2020 May 7;6(1):38).