DYRK (dual-specificity tyrosine-regulated kinases) are an evolutionary conserved family of protein kinases with members from yeast to humans. In humans, DYRKs are pleiotropic factors that phosphorylate a broad set of proteins involved in many different cellular processes, including those associated with all the hallmarks of cancer. In accordance with an involvement of DYRK kinases in the regulation of tumorigenic processes, an increasing number of research studies are showing either alterations of DYRK gene expression in tumor samples and/or providing evidence of DYRK-dependent mechanisms that contribute to tumor initiation and/or progression.
- DYRK kinases
- cellular signaling
- expression dysregulation
- cell cycle
- cell survival
- tumor progression
- kinase inhibitors
1. The DYRK Family of Kinases
The first cancer gene identified, the proto-oncogene c-Src, was found to encode a protein kinase [1]. Yet, since then, almost a hundred kinase genes have been attributed a tumor suppressor or oncogenic role, and they represent the most abundant class of cancer driver genes known to date [2]. Dual-specificity tyrosine-regulated kinases (DYRKs) belong to the CMGC group of kinases, which includes cyclin-dependent kinases (CDKs), mitogen-activated protein kinases (MAPKs), CDK-like kinases, the serine-arginine-rich protein kinase, Cdc2-like kinases (CLKs) and members of the RCK family [3]. The DYRK family is formed by three subfamilies: the DYRK subfamily, the homeodomain-interacting kinases (HIPKs), and the pre-messenger RNA-processing protein 4 kinases (PRP4Ks) [3]. Here, we will use “DYRK” to refer specifically to the DYRK subfamily, which contains five members in humans that are clustered into two classes based on their phylogenetic relationships [4]: class I DYRKs, DYRK1A and DYRK1B (also known as Mirk from minibrain-related kinase) and class II DYRKs, DYRK2, DYRK3 (also known as REDK from regulatory erythroid kinase) and DYRK4 (Figure 1A). DYRK kinases phosphorylate a broad set of substrates that are involved in a wide range of cellular processes, and they are thought to fulfill essential biological functions both during development and in maintaining homeostasis during the adult life [4,43,94,126,127]. Consequently, the aberrant regulation or expression of DYRK kinases has been associated with several human pathologies, including cancer.
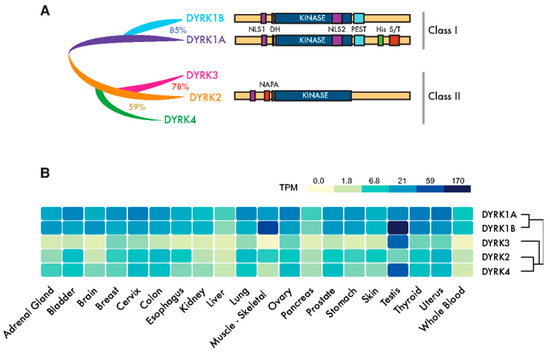
Figure 1. Dual-specificity tyrosine-regulated kinase (DYRK) protein kinases: primary structure and expression. (A) Scheme of the mammalian family of DYRKs, indicating their phylogenic relationships, degree of homology and protein domains. The catalytic domain (KINASE) and the DYRK homology box (DH) are common to all members of the family. Class I DYRKs have two nuclear localization signals (NLSs) (NLS1 and NLS2) and a proline-, glutamic acid-, serine- and threonine-rich (PEST) motif. DYRK1A also includes a tract of 13 consecutive histidine residues (His) and a region enriched in serine/threonine residues (S/T) at the C-terminus. Class II DYRKs have a common structure, with the characteristic N-terminal autophosphorylation accessory (NAPA) domain at the N-terminus. In the case of DYRK2 and DYRK4, functional NLSs have been described within the noncatalytic N-terminus. (B) The expression of human DYRKs based on the Genotype-Tissue Expression (GTEx) data represented as the median TPMs (transcripts per million: GTEx Analysis Release V8, www.gtexportal.org/home, dbGaP Accession phs000424.v8.p2; brain: cortex; cervix: ectocervix; colon: sigmoid colon; esophagus; mucosa, kidney: cortex; skin: suprapubic—not sun-exposed).
The members of the DYRK family all share a highly conserved catalytic domain with special features within the CMGC group [5] [5] and the so-called DYRK homology (DH) box motif located upstream of it (Figure 1A). In addition, DYRK kinases present class-specific domains: DYRK1A and DYRK1B harbor a proline-, glutamic acid-, serine- and threonine-rich (PEST) motif in the noncatalytic C-terminal region and equally positioned nuclear localization signals (NLS) (Figure 1A). On the other hand, class II DYRKs present a N-terminal autophosphorylation accessory region (NAPA) domain, essential for catalytic activation [6] [6] (Figure 1A). All human DYRKs accumulate in the cytosol of cells, and DYRK1A, DYRK2 and DYRK4 can be imported into the nucleus by means of dedicated NLSs [7][8][9][7,8,9]. Nuclear DYRK1A is a chromatin-associated kinase capable of regulating the gene expression [10][11] [10,11], and it is functionally linked to the DNA damage response (DDR) [12][13][14][12,13,14]. Chromatin association in the DDR context has also been described for DYRK1B [15]. Moreover, a DYRK1A-specific run of histidine residues targets this family member to the subnuclear splicing compartment [7], and the noncatalytic N-terminal domain of DYRK3 serves to localize it to stress granules [16]. Both the histidine run in DYRK1A and the N-terminus of DYRK3 participate in the generation of phase-separated subcellular compartments [17][18][17,18]. Changes in the subcellular localization of DYRK proteins have been observed in response to different signals, such as that of DYRK2 in response to DNA damage or proinflammatory signals [19][20][19,20] or DYRK1A in response to Wnt signaling [21]. However, how the subcellular localization of DYRKs is regulated or how it contributes to their activity is still not well-understood.
DYRK1A and DYRK1B are expressed ubiquitously in human tissues, whereas class II DYRKs are generally expressed more weakly and in a more tissue-restricted pattern (Figure 1B). The expression of DYRKs is regulated through alternative promoters that generate transcripts with distinct 5′-untranslated regions and/or encoding different N-terminal regions [4]. In addition, alternative splicing generates multiple protein isoforms of unclear functional significance [4][8][22][23][24][4,8,22,23,24]. DYRKs are also subject to other post-transcriptional events, such as microRNAs-mediated gene silencing [25][26][27] [25,26,27] or local translation [28].
DYRK kinases are “dual specificity” kinases, as they can phosphorylate both tyrosine (Y) and serine/threonine (S/T) residues, although Y-phosphorylation is limited to their autophosphorylation activity [29]. These kinases are activated by the phosphorylation of residues within the activation loop, which drives a conformational switch from the inactive to active state [30][31][30,31]. Unlike other kinase families, this key event in DYRKs is an autocatalytic reaction that occurs during protein synthesis and that generates a constitutively active kinase [32]. As DYRK activation does not depend on upstream kinases, other regulatory mechanisms are thought to operate, including the dephosphorylation of residues in the activation loop, allosteric phosphorylation performed by other kinases [9][33][34][35][36][9,33,34,35,36], interactions with scaffolding proteins [37][38][39][37,38,39], accessibility to substrates due to changes in the subcellular localization or regulation of protein levels at transcriptional and/or post-transcriptional levels.
2. The Role of DYRKs in Cancer
DYRKs phosphorylate a wide range of substrates, including factors associated with one or several of the hallmarks of cancer [40] [40] (Figure 2). Of all the DYRKs, only DYRK1A has been identified in high-throughput cancer studies, initially as a potential tumor suppressor using Tumor Suppressor and Oncogene Explorer (TUSON) [41], and later on, it was proposed as a driver in liver cancer through a study that identified such drivers according to mutations in unusual nucleotide contexts [42]. Although these results would suggest that DYRKs are not major drivers of cancer, further evidence that they play a role in oncogenic processes has emerged over the past two decades, either by detecting alterations to the DYRK expression in tumor tissues, and/or by analyzing the impact of DYRK-dependent phosphorylation on substrates involved in cancer-related events (recently reviewed in XX).
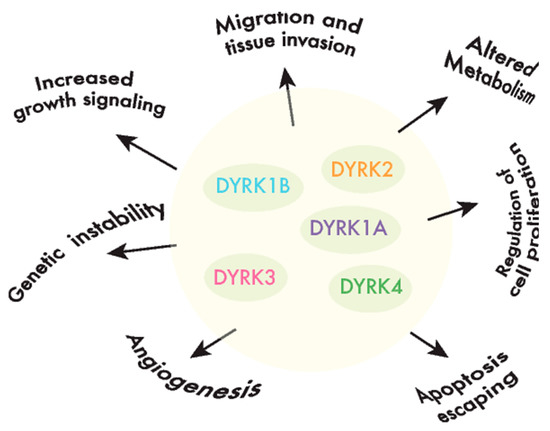
Figure 2. DYRKs are involved in cancer-associated processes. DYRK kinases participate in the regulation of crucial cell events, the perturbation of which is responsible for producing important features in cancer cells or the hallmarks of cancer.
DYRK1A is the most extensively studied member of the family, mainly due to its key role in neurogenesis and in the etiology of some of the pathological traits associated to Down syndrome (recently reviewed in [43]). In addition, DYRK1A haploinsufficiency caused by de novo truncations or by missense-inactivating mutations underlies a rare, severe disorder, the DYRK1A haploinsufficiency syndrome (also known as MRD7 or Mental Retardation, Autosomal Dominant 7: OMIM#614104 and ORPHA:464311 and 268261; [44][45][44,45] and references therein). Regarding a putative role in cancer, several studies have ascribed opposite functions to this family member [46][47][48][49][50][55,66,68,79,85], reflecting a very complex scenario. Therefore, it remains unclear as to whether DYRK1A acts as a tumor suppressor or a tumor promoter or, more probably, as either, depending on the tumor context. Unlike DYRK1A, findings on its closest paralog DYRK1B point mostly to a prosurvival and protumorigenic role (recently reviewed in [51][94]). DYRK2 activity appears to affect crucial processes linked to tumor progression like the cell cycle, the DDR, epithelial-to-mesenchymal transition, the xenobiotic response system and cellular proteostasis (recently reviewed in [52][53][126,127]). Alterations in the expression of DYRK2 have been observed in different types of cancer, and in general, the weaker the expression of DYRK2, the worse the prognosis [54][55][56][57][58][59][60][61][137-140,142–145]; in addition, DYRK2 gene silencing confers an enhanced proliferative capacity and metastatic potential in vivo [56][61][62][139,145,146]. However, some discordant phenotypes have been described when studying breast cancer [63][64][65][66][56,134,147,148]. Finally, the contribution of DYRK3 and DYRK4 to tumorigenesis is less clear, with very little evidence for the participation of DYRK3 [167] and no evidence for that of DYRK4.
3. DYRK Inhibitors as Antitumor Therapies
Chemical compounds that bind and functionally block protein kinases have been studied extensively and employed as antitumor agents, both in research and in clinical trials. Although the role of DYRK family members in tumorigenesis and tumor progression has not been fully elucidated, pharmacological inhibitors of DYRK kinases have been tested in laboratories for their antimalignant activity, and a few of them are already undergoing clinical trials.
In the case of DYRK1A, the search for both naturally occurring and synthetic inhibitors has been extensive given that DYRK1A may be a potential pharmacological target not only in cancer but, also, in neurodegenerative diseases (reviewed in [43]), Down syndrome [67][68][69][70] [169,170,171,172] and diabetes (reviewed in [71][173]). Comprehensive reviews on DYRK1A inhibitors can be found at [72][73][74][174,175,176]. Compounds targeting DYRK1B, with either restricted or broad specificity, have been used as research tools, and they display toxicity towards several types of cancer cells or they promote the cell cycle re-entry of quiescent tumor cells (reviewed in [51][94]), with positive effects in combinatorial drug approaches. For instance, the DYRK1B inhibitor AZ191 [75][52] increases the anticancer effects of doxorubicin in liposarcoma cell lines [76][96] or sensitizes pancreatic adenocarcinoma cell lines to mTOR inhibition [77][115]. However, AZ191 has been also shown to counteract the antitumor effects of the lysosome inhibitor Bafilomycin A1 in hepatocellular carcinoma cell lines [78][111]. For DYRK2, experimental data on the antitumor effects of the natural DYRK2 inhibitor curcumin and of the synthetic compound LDN192960 was obtained in both in vitro and in vivo models of triple negative breast cancer (TNBC) and multiple myeloma, supporting the hypothesis that DYRK2 is a promising pharmaceutical target in these malignancies [64][79][134,162]. Finally, better understanding the role of DYRKs in tumor cells has proven valuable by helping to identify combinatorial therapeutic approaches, as in the cases of the DYRK1B inhibitors that enhance the inhibitory efficiency of MEK and mTOR [80][81][82] [107,109,187] or DYRK2 inhibition sensitizing MDA-MB-468 cells to the proteasome inhibitor bortezomib [66][148].
The only inhibitors of the DYRK family members currently being screened in clinical trials were identified as inhibitors of other protein kinases. In particular, compound CX-4945 was initially identified as a casein kinase 2 inhibitor, but it was subsequently shown to be a potent DYRK1A and DYRK1B inhibitor [69][171], and it is currently in phase I and II clinical studies for medulloblastoma, cholangiocarcinoma and basal cell carcinoma (NCT02128282, NCT03904862 and NCT03897036). Compound OTS167, a chemical initially described as a maternal embryonic leucine zipper kinase inhibitor, has potent anti-DYRK1A activity [83][189], and it is currently being assessed in clinical trials for the treatment of advanced breast cancer and TNBC (phase I) and for multiple types of leukemia (phase II: NCT02795520). Finally, two other DYRK inhibitors have been assessed in clinical trials for non-neoplastic disorders: GSK-626616 [16] [16] completed a phase I clinical trial to evaluate its action on anemia (NCT00443170), and lorecivivint, a potent CLK2 inhibitor that also inhibits DYRK1A [84][190], is being studied in a phase II trial for the treatment of moderate-to-severe symptomatic osteoarthritis (NCT03706521). Thus, they could be repurposed in trials for the treatment of specific cancer types.
4. Conclusions
In the last decade, more experimental evidence indicates that DYRK protein kinases are a novel class of “kinase-of-interest” in cancer. However, this evidence mostly comes from studies exploring DYRK expressions in tumor tissues and/or the phenotypic changes triggered by manipulating the DYRK protein levels in cancer cell lines. These data not only provide a partial and confusing picture of the influence of DYRKs in tumor initiation and progression, but also, they highlight the many questions that still need to be addressed. In particular, it remains unclear which molecular pathways are regulated by DYRKs in different tumor types and which of them selectively trigger cells to engage in neoplastic transformation or enhance the malignant phenotype of tumor cells. Resolving these issues will not only help understand the biology behind the activity of these kinases, but also, it will provide a basis for the rational design of therapeutic approaches based on inhibitors. In this regard, while incomplete, the currently available data provides precious information on which forthcoming therapeutic approaches may be based. Therefore, the tumor types in which downregulation of the DYRK kinase has been associated with increased tumor growth and/or invasiveness should not be considered for trials with DYRK inhibitors. Conversely, inhibitors targeting DYRK family members that are known to favor the tumorigenesis of specific tumor types should be considered for such trials. Nevertheless, putative side effects due to the inhibition of members that are essential to maintaining cellular homeostasis in normal cells, such as the dosage-sensitive DYRK1A or DYRK1B, should be carefully monitored. In this context, engineering drugs to increase their specificity, exclusively targeting proliferating cells, would be worthwhile. Finally, and considering the differential and sometimes opposite roles of distinct DYRK kinases in tumor progression, selectivity towards a specific member of the family is crucial and, at the same time, very challenging, particularly given the strong structural similarity of the catalytic domain. Smart solutions might include an allosteric drug design or other additional efforts to increase compound selectivity.
To conclude, many important advances in understanding how the dysregulation of DYRK protein kinases is associated to pathological phenotypes in humans have been made in recent years—in particular, in terms of their involvement in cancer. Still, many secrets behind the oncogenic or protective potential of DYRK kinases remain to be revealed, and we anticipate that the field will continue to grow for the foreseeable future.