Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Krasimir Kostov and Version 2 by Rita Xu.
Hypertension (HTN) is one of the most prevalent diseases worldwide and is among the most important risk factors for cardiovascular and cerebrovascular complications. It is currently thought to be the result of disturbances in a number of neural, renal, hormonal, and vascular mechanisms regulating blood pressure (BP), so crucial importance is given to the imbalance of a number of vasoactive factors produced by the endothelium.
- endothelin-1
- hypertension
- oxidative stress
- low-grade inflammation
- endothelial dysfunction
- arterial stiffness
1. Introduction
Hypertension (HTN) is one of the most prevalent socially significant diseases and is among the most important preventable risk factors for other diseases [1]. The heart, brain, kidneys, and peripheral arteries are often affected, which is a cause of early disability and reduced life expectancy in patients [2]. This necessitates that the prevention and treatment of HTN be among the top priorities of public health worldwide [3].
HTN is a heterogeneous disease with a complex pathogenesis. It is currently thought to be the result of disturbances in a number of neural, renal, hormonal, and vascular mechanisms regulating blood pressure (BP) [4], as crucial importance is given to the imbalance of a number of vasoactive substances, some of which are produced from the vascular endothelium [5]. The endothelium responds to humoral, neural, and especially hemodynamic stimuli, and regulates platelet function, inflammatory responses, growth and migration of vascular smooth muscle cells (VSMCs), and changes in the structure of the vascular extracellular matrix [6][7][6,7]. In addition to these functions, it modulates vascular tone by synthesizing and releasing a number of vasoactive factors that may have vasodilatory effects, such as nitric oxide (NO), prostacyclin (PGI2), and endothelium-derived hyperpolarizing factor, and vasoconstrictor effects, such as thromboxane A2 and endothelin-1 (ET-1) [8]. In HTN, the delicate balance between vasodilators and vasoconstrictors is disturbed, leading to endothelial dysfunction (ED) with excessive release of vasoconstrictor substances, such as ET-1 [9][10][9,10].
ET-1 was first isolated in 1988 by Yanagisawa and colleagues from the culture supernatant of porcine aortic endothelial cells (ECs). It is composed of 21 amino acids and two intrachain disulfide linkages in the molecule [11]. Shortly after the discovery of ET-1, two other structurally similar isopeptides, named ET-2 and ET-3, were isolated [12]. ET-1 is the predominant isopeptide involved in regulating the cardiovascular system, and vascular ECs are the most abundant source of ET-1. In addition to ECs, ET-1 is expressed in a wide variety of cells including VSMCs, cardiomyocytes, fibroblasts, macrophages, epithelial cells in the lungs and kidneys, neurons, and glial cells [13]. The endothelins (ETs) are produced from their corresponding approximately 200-residue prepropolypeptides that are encoded by three distinct genes. These peptides are converted into inactive 38- or 39-amino acid intermediates called Big ETs (Big ET-1, Big ET-2, and Big ET-3) by furin-like endopeptidase. The Big ETs are then activated via proteolytic cleavage by the ET-converting enzymes (ECEs), ECE-1 and ECE-2 [14] (Figure 1).
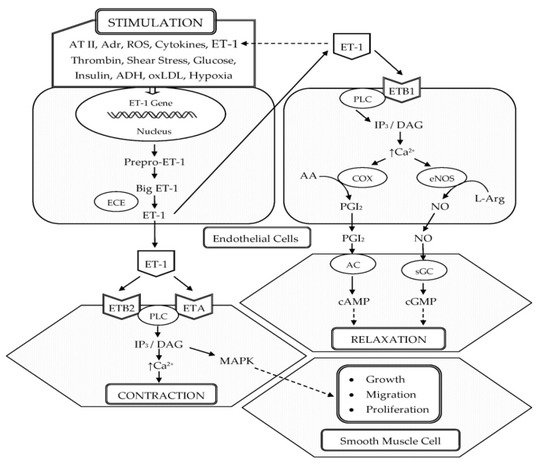
Figure 1. Schematic representation of the biosynthesis and vascular effects of ET-1 in endothelial and smooth muscle cells. ET-1 is generated by ECs in response to different stimuli. ET-1 mediates vasoconstriction by activating ETA and ETB2 receptors on VSMCs. Vasodilation by ET-1 is mediated through the activation of ETB1 on ECs, which increases the production of NO and PGI2. In addition, ET-1 is a potent mitogen that simulates the growth, proliferation, and migration of VSMCs. Abbreviations: ET-1, endothelin-1; AT II, angiotensin II; Adr, adrenaline; ROS, reactive oxygen species; ADH, antidiuretic hormone; oxLDL, oxidized low-density lipoproteins; ECE, endothelin-converting enzyme; ETA, endothelin receptor subtype A; ETB1, endothelin receptor subtype B1; ETB2 endothelin receptor subtype B2; PLC, phospholipase C; IP3, inositol trisphosphate; DAG, diacylglycerol; Ca2+, calcium ions; COX, cyclooxygenase; AA, arachidonic acid; PGI2, prostacyclin; eNOS, endothelial nitric oxide synthase; L-Arg, L-arginine; NO, nitric oxide; AC, adenylate cyclase; cAMP, cyclic adenosine monophosphate; sGC, soluble guanylate cyclase; cGMP, cyclic guanosine monophosphate; MAPK, mitogen-activated protein kinase; ↑, increased.
In the vasculature, ET-1 acts on ETA and ETB (ETB1 and ETB2) receptors located on the VSMCs and ECs to induce vascular contraction or vasodilation [15]. Vasoconstrictive action of ET-1 is mainly mediated through ETA. ET-1–ETA interaction on VSMCs increases intracellular calcium, leading to the phosphorylation and activation of myosin light chain, which causes vasoconstriction [16][17][16,17]. In pathophysiological conditions, the expression of ETB2 on VSMCs is increased and ET-1–ETB2 interaction also promotes vasoconstriction [16]. ET-1 induces long-lasting vasoconstriction resulting from the slow dissociation rate from ET receptors [18]. In 40% of adults, a genetically prohypertensive phenotype is present or is the result of partially epigenetically mediated environmental effects. This is related to the predominance of vasoconstrictor actions of ET-1 mediated by ETA and ETB2 in VSMC, which is due to the hypertensive effect of increased endothelial expression of the ET-1 gene (EDN1) [19]. However, vasodilation by ET-1 is mediated through ETB1 on ECs, which increases the production of NO and PGI2 [17] (Figure 1).
ET-1 is continuously released from the endothelial constitutive pathway. Low levels of ET-1 promote vasodilatation, whereas higher and pathophysiological concentrations increase BP and total peripheral vascular resistance [13]. In healthy volunteers, low doses of ET-1 infused into the brachial artery cause vasodilatation, consistent with ETB-mediated release of vasodilators, but this was followed by sustained vasoconstriction of the forearm vascular bed at higher doses because the peptide accessed the smooth muscle ETA receptors [20]. Thus, ETB-mediated release of NO and other vasodilators is crucial in acting as a counterregulatory pathway to limit ETA-mediated vasoconstriction. In pathophysiological conditions where there is ED with a loss of vasodilators, the vasoconstrictor and other pathophysiological effects of ET-1, such as cell proliferation, will be potentiated [13]. In patients with essential HTN, the activity of exogenous ET-1 is increased, similar, or decreased compared to normotensive subjects, depending on which vascular district or scheme of administration is considered [21].
2. Data on the Participation of ET-1 in the Development of HTN
Due to its ability to maintain basal vascular tone [22][23][22,23] and homeostasis of sodium and water [24][25][24,25], it is suggested that ET-1 is involved in some forms of HTN [26], which is supported by various experimental and clinical observations.
2.1. ET-1 in Experimental HTN
The role of ET-1 in HTN was initially observed in models of experimental HTN, and more recently by using genetically modified mouse models in which a component of the endothelin system was either knockdown or overexpressed in certain specific organs or tissues [27]. It should be noted that when ET-1 is overexpressed in the endothelium of transgenic mice, BP is significantly higher than in wild-type mice [28]. Moreover, selective knockout of ET-1 in the collecting duct of the nephron was associated with a higher BP and the development of salt-sensitive HTN [29]. A similar BP phenotype was obtained with the deletion of ETB or both ETA/ETB receptors in collecting duct cells, suggesting the important role of the renal endothelin system in the development of salt-sensitive HTN [30][31][30,31]. In non-transgenic animals, elevated ET-1 production was found in salt-sensitive and some other models of experimental HTN, such as deoxycorticosterone (DOCA)-salt HTN, DOCA-salt-treated spontaneously hypertensive rats (SHR), and Dahl salt-sensitive rats, 1-kidney 1 clip (1K1C) Goldblatt hypertensive rats, SHR 2-kidney 1 clip (2K1C) Goldblatt hypertensive rats, angiotensin II-infused rats, and stroke-prone SHR [32]. In the models known to overexpress vascular ET-1, BP significantly decreases upon the administration of selective ETA or mixed ETA/ETB receptor antagonists [33].
2.2. ET-1 in Human HTN
One of the first comparisons of ET-1 concentrations in people with HTN was made between pheochromocytoma patients and healthy controls. Higher levels of ET-1 were observed in patients with pheochromocytoma. In this report, the authors note that HTN in patients with pheochromocytoma is mainly catecholamine dependent, but may be secondarily ET-1 dependent [34]. These data are supported by previously reported cases in patients with hemangioendothelioma who have significantly elevated ET-1 levels along with HTN [35]. Elevated ET-1 levels and high BP in patients from these studies returned to normal after surgical removal of the tumors [34][35][34,35]. In addition, resistant HTN with elevated ET-1 levels has been observed more frequently in patients of African-American descent or those with obesity, in whom the risk of developing cardiovascular and renal diseases is increased [36]. Furthermore, in individuals with normal BP, high plasma ET-1 levels are associated with the development of HTN [37]. The role of ET-1 in the development of the hypertensive process is also supported by data in patients with essential HTN or resistant HTN, which show that when treated with a nonselective ET-receptor antagonist bosentan [38] or with the selective ETA receptor antagonist darusentan [39][40][41][39,40,41], BP is significantly reduced. These data are also consistent with the meta-analyses, which show that HTN patients have higher plasma concentrations of ET-1 than control subjects [42]. Other authors have reported that the levels of ET-1 are normal in patients with essential HTN, but point out that the local levels of ET-1 in the vascular wall are elevated [36][43][44][36,43,44]. The controversial and not always consistent results regarding ET-1 concentrations in patients with HTN are probably related to two main reasons. The first is that its elimination from the blood is too fast (plasma half-life 1–2 min) [45]. The second is that the secretion of ET-1 by ECs is polarized mainly to the underlying VSMCs, leading to a minimal increase in its circulating levels [46]. Other possible causes of these disparate results are the specificity of the antibodies used in the immunoassay, the degree of cardiovascular damage, dietary salt intake, obesity, diabetes, and race [24]. All of the above findings support the hypothesis that ET-1 may have an important pathogenetic role in the development of HTN (Table 1).
Table 1.
Studies on the contribution of ET-1 to the hypertensive phenotype in humans.
| Study | Results | Significance | |
|---|---|---|---|
| Saito, 1990 [47] |
Patients with essential HTN showed a significant elevation in the plasma ET-1 level compared with age-matched control subjects. | p | < 0.01 |
| Shichiri, 1990 [48] |
Patients with essential HTN had significantly higher plasma ET-1 levels than normal subjects. | p | < 0.025 |
| Oishi, 1994 [34] |
In patients with pheochromocytoma, the hypertensive group had higher ET-1 than the normotensive group. Elevated plasma ET-1 concentrations returned to normal levels after surgical resection of the tumor. | Higher, but NS | |
| Parrinello, 1996 [49] |
ET-1 levels were significantly higher in obese hypertensives and obese normotensives than in lean normotensives. In addition, ET-1 levels were significantly higher in obese hypertensives than in obese normotensives. | p | < 0.05 |
| Amoroso, 1996 [50] |
Patients with HTN had significantly higher plasma ET-1 concentration than normal subjects. | p | < 0.02 |
| Schneider, 2000 [51] |
Basal ET-1 was significantly higher in hypertensive than in normotensive subjects, both in venous and arterial samples. There was no significant difference between venous and arterial ET-1 concentrations. | p | < 0.01 |
| Parissis, 2001 [52] |
Patients with HTN showed significantly higher levels of ET-1 compared with normotensive controls. | p | < 0.01 |
| Kostov, 2014 [53] |
Serum levels of ET-1 are significantly higher in patients with mild and severe HTN compared to the control group. | p | < 0.02 |
| Gu, 2015 [54] |
Plasma ET-1 levels were higher in hypertensives than in controls. | p | < 0.001 |
| Kostov, 2016 [55] |
Serum ET-1 concentrations were significantly higher in hypertensive patients with type 2 diabetes than in prehypertensive patients with diabetes and healthy normotensive controls. | p | < 0.05 |
Abbreviations: ET-1, endothelin-1; HTN, hypertension; NS, not significant.