Lipid rafts are dynamic assemblies of glycosphingolipids, sphingomyelin, cholesterol, and specific proteins which are stabilized into platforms involved in the regulation of vital cellular processes. Recent reports have demonstrated that lipid rafts are spatially and compositionally heterogeneous in the single-cell membrane. Sphingomyelin-rich rafts that are spatially and functionally distinct from the cholesterol-rich rafts are found at spreading platelets. Fibrin is translocated to sphingomyelin-rich rafts and platelet sphingomyelin-rich rafts act as platforms where extracellular fibrin and intracellular actomyosin join to promote clot retraction.
- lipid rafts
- detergent-resistant membrane
- heterogeneity
- platelets
Lipid rafts are dynamic assemblies of glycosphingolipids, sphingomyelin, cholesterol, and specific proteins which are stabilized into platforms involved in the regulation of vital cellular processes. The rafts at the cell surface play important functions in signal transduction. Recent reports have demonstrated that lipid rafts are spatially and compositionally heterogeneous in the single-cell membrane. In this review, we summarize our recent data on living platelets using two specific probes of raft components: lysenin as a probe of sphingomyelin-rich rafts and BCθ as a probe of cholesterol-rich rafts.
1. Platelet Lipid Rafts
- Platelet Lipid Rafts
The fluid mosaic model has supported our understanding of cellular membranes for a long time. Recent studies suggest that plasma membrane lipids are not homogeneously distributed and that the membranes may contain microdomains or compartments. Glycosphingolipids form microdomains containing cholesterol in the cell membrane. Glycosphingolipid- and cholesterol-rich microdomains are referred to as lipid rafts. Lipid rafts are dynamic assemblies of glycosphingolipids, sphingomyelin, cholesterol, and proteins which are stabilized into platforms involved in the regulation of a number of cellular processes [1]. Lipid rafts are isolated as a detergent-resistant membrane (DRM) fraction by sucrose density gradient centrifugation. Recent studies have demonstrated that lipid rafts are spatially and compositionally heterogeneous in the cell membrane. In migrating T cells, GM3 ganglioside-rich rafts containing a chemokine receptor are present at their leading edge, whereas GM1-rich rafts containing integrin β1 are present at their uropod [2].
In 1996, platelet DRM was shown to be rich in glycoprotein CD36, Src, and Lyn [3]. Platelet rafts are important membrane microdomains in responses such as adhesion and aggregation. The localization of the adhesion receptor glycoprotein (GP)Ib-IX-V complex to lipid rafts is required for platelet adhesion to the vessel wall by binding the von Willebrand factor (vWF) [4][5][4,5]. In resting platelets, phosphatidylserine (PS) is asymmetrically restricted to the inner leaflet of the plasma membrane. An increase in intracellular Ca2+ concentration during platelet activation can lead to the exposure of PS in the outer leaflet. PS forms a procoagulant binding site for tenase and prothrombinase coagulation complexes. Lipid rafts are required for the release of PS-exposing extracellular vesicles from platelets [6]. Thus, lipid rafts are critical membrane domains in platelet activation processes [7][8][7,8]. Interestingly, platelet DRM shifts to a higher density in sucrose gradients upon thrombin receptor activating peptide (TRAP) stimulation [9]. Trace amounts of actin are observed in rafts from resting platelets, but a marked increase in the amount of actin is found in rafts upon platelet stimulation by TRAP. Platelet DRM also shifts to a higher density in sucrose gradients upon adenosine diphosphate (ADP) stimulation [10].
A protease-nicked and biotinylated derivative (BCθ) of perfringolysin O (θ-toxin) binds specifically to cholesterol-rich microdomains of intact cells [11]. In resting platelets, BCθ-positive cholesterol-rich rafts are uniformly distributed on the cell surface. Upon interaction with fibrinogen, BCθ-positive cholesterol-rich rafts accumulate at the tips of filopodia and at the leading edge of spreading platelets [12]. The adhesion-dependent raft aggregation is accompanied by the concentration of the tyrosine kinase c-Src and the tetraspanin CD63 in cholesterol-rich rafts. The perfringolysin O derivative BCθ recognizes a subpopulation (cholesterol-rich rafts) of platelet DRM rafts, suggesting that a heterogeneous population of lipid rafts exists in platelets [11]. However, little is known about raft heterogeneity in platelet membranes.
2. Sphingomyelin-Rich Rafts of Platelets
- Sphingomyelin-Rich Rafts of Platelets
We have been identifying glycosphingolipid-binding proteins [13][14][15][16][17][18][19][13–19] and investigated the signal transduction in lipid rafts of platelets [20]. Previously, we reported that clot retraction is mediated by the coagulation factor XIII (FXIII)-dependent fibrin-integrin αIIbβ3-myosin axis in platelet sphingomyelin (SM)-rich membrane rafts [21]. Clot retraction is a process driven by outside-in signaling by the platelet integrin αIIbβ3, resulting in the contraction of the fibrin mesh and the formation of mechanically stable thrombi. To elucidate the function of platelet lipid rafts, we identified DRM-raft-specific proteins from activated platelets. We isolated the DRM raft fraction of platelets treated with thrombin by sucrose gradient centrifugation. Several specific proteins were present in the DRM fraction of thrombin-stimulated platelets. By mass spectrometry, we identified three proteins of 65, 50, and 47 kDa as fibrins α, β, and γ, respectively. These findings were supported by the results of immunoblot analysis using an anti-fibrinogen/fibrin polyclonal antibody. In resting platelets, fibrinogens Aα (67 kDa), Bβ (52 kDa), and γ (47 kDa) were present in the non-raft fraction. In contrast, fibrins α (65 kDa), β (50 kDa), and γ (47 kDa) were exclusively present in the DRM fraction of platelets treated with thrombin (Figure 1A) [21]. Therefore, we investigated the subcellular distribution of fibrin and BCθ-positive cholesterol-rich rafts on thrombin-stimulated spreading platelets by scanning immunoelectron microscopy. Fibrin was localized in the central area of spreading platelet (Figure 1B, left panel). In contrast, BCθ-positive cholesterol-rich rafts were localized evenly on the membrane (Figure 1B, right panel). These observations suggest that fibrin is translocated to platelet rafts other than cholesterol-rich rafts following thrombin stimulation.
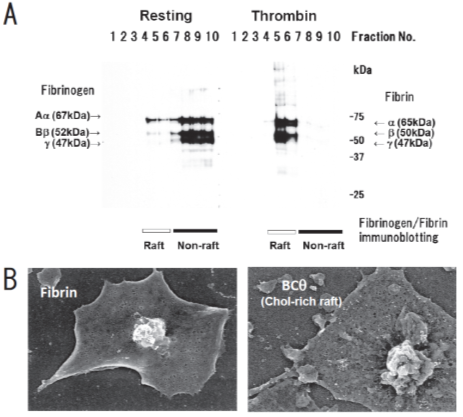
Figure 1. Fibrin translocation to lipid rafts in central region of spreading platelets stimulated with thrombin. (A) Sucrose density gradient analysis of washed human platelets. Resting platelets (left) and platelets stimulated for 3 min with 1 U/mL thrombin (right) were lysed in Triton X-100 and then adjusted to 40% sucrose. A sucrose gradient (5–30%) in a volume of 6 mL was layered over the lysate (4 mL) and was centrifuged. Ten fractions were collected from top to bottom after centrifugation and subjected to immunoblotting with an anti-fibrinogen polyclonal antibody. In resting platelets, fibrinogens Aα (67 kDa), Bβ (52 kDa), and γ (47 kDa) were detected in the non-raft fraction (lanes 7–10). In contrast, fibrins α (65 kDa), β (50 kDa), and γ (47 kDa) were detected in the raft fraction (lanes 5,6) of thrombin-stimulated platelets. (B) Localization of fibrin (left panel) and BCθ-positive cholesterol-rich rafts (right panel) of thrombin-stimulated spreading platelets on fibronectin by scanning immunoelectron microscopy. Spreading platelets were incubated with 15 μg/mL BCθ for 30 min followed by glutaraldehyde fixation and immunolabeling with anti-biotin IgG gold. Gold-positive fibrins were localized in the central region of spreading platelet (left). In contrast, gold-positive cholesterol-rich rafts were localized uniformly on the membrane (right). The study was approved by the institutional ethics committee.
Lysenin, the earthworm toxin, is a specific probe of sphingomyelin (SM)-rich rafts in living cells [22][23][22,23]. SM is a major component of raft lipids in platelets [9]. Therefore, we investigated the subcellular distribution of SM-rich rafts in spreading platelets. Lysenin-positive SM-rich rafts were localized in the central area of adhering platelets stimulated with thrombin (Figure 2A, left panel). Lysenin-positive SM-rich rafts and fibrin mostly colocalized as a patch in the double-stained the central area of spreading platelets stimulated with thrombin (Figure 2A, middle panel). Next, we investigated the spreading of platelets by time-lapse differential interference contrast (DIC) imaging (Figure 2B) and lysenin staining (Figure 2C). In resting platelets (Figure 2C, 0 min), lysenin-positive SM-rich rafts were uniformly distributed on the cell surface. At an early stage of the spreading of platelets treated with thrombin for 3 min, SM-rich rafts were mainly localized in the central area of adhering platelets with some distributed in the lamellipodia. At a late stage of spreading of platelets treated with thrombin for 15 min, almost all SM-rich rafts were in the central area. Furthermore, we also demonstrated the translocation of myosin to the DRM raft fraction following thrombin stimulation and the colocalization of activated myosin with fibrin in SM-rich rafts of adhering platelets stimulated with thrombin [21]. These observations suggest that SM-rich rafts act as platforms of fibrin-mediated outside-in signaling, leading to clot retraction. To support this idea, the clot retraction of SM-depleted platelets from SM synthase 1 and SM synthase 2 knockout mice was delayed significantly. As a result, we demonstrated that fibrin converted by thrombin translocates immediately into platelet DRM rafts in a coagulation factor XIII (FXIII)-dependent manner. Therefore, we proposed that fibrin is translocated to SM-rich rafts in the presence of FXIII crosslinking activity and that platelet SM-rich rafts act as platforms where extracellular fibrin and intracellular actomyosin join to promote clot retraction [21][24][25][21,24,25]. A spatial distinction between SM-rich rafts and cholesterol-rich rafts in platelets is illustrated (Figure 3).
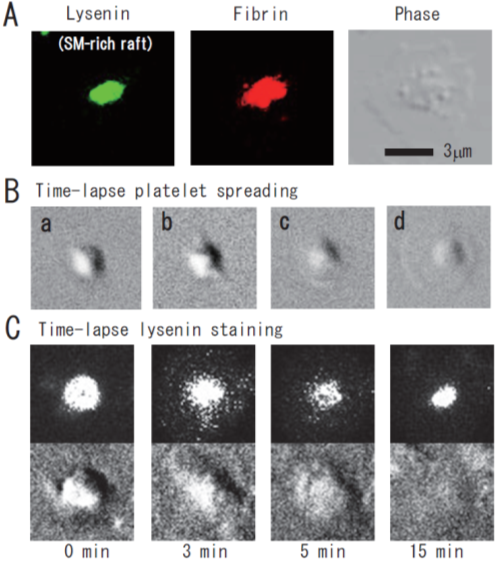
Figure 2. Immunocytochemical colocalization of fibrin with sphingomyelin-rich rafts in central region of spreading platelets stimulated with thrombin. (A) Immunocytochemical colocalization of fibrin with sphingomyelin-rich rafts in central region of thrombin-stimulated spreading platelets. Green fluorescent protein (GFP)–lysenin-positive sphingomyelin-rich rafts (left panel). Alexa 594-labeled fibrin (middle panel). Phase contrast (right panel). Scale bar, 3 μm. (B) Time-lapse platelet spreading after thrombin stimulation on fibronectin-coated glass strip. (a) 0 min; (b) 0.2 min, filopodia formation; (c) 2 min, spreading; (d) 10 min, complete spreading. (C) Time-lapse lysenin-positive sphingomyelin-rich raft staining. Washed platelets were incubated with GFP-lysenin for 10 min and then stimulated with 1 U/mL thrombin. The time-lapse fluorescent and DIC images were captured using Olympus LCV110.
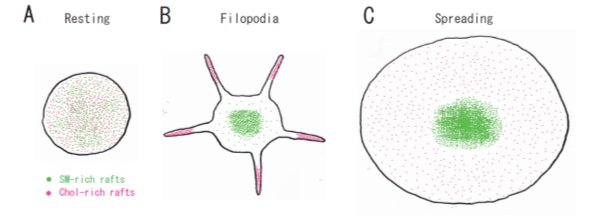
Figure 3. A spatial distinction between SM-rich rafts and cholesterol-rich rafts in platelets. (A) In resting platelets, SM-rich rafts (green) and cholesterol-rich rafts (red) are uniformly distributed on the cell surface. No spatial distinction is observed by confocal microscopy. (B) Cholesterol-rich rafts accumulate at the tips of filopodia of adhering platelets [12]. SM-rich rafts are mainly localized in the central area of adhering platelets with some distributed in the lamellipodia. (C) SM-rich rafts are in the central area of full spreading platelets. Cholesterol-rich rafts were localized evenly on the membrane [21].
3. Raft Heterogeneity
- Raft Heterogeneity
Platelet DRM shifts to a higher density in sucrose gradients upon platelet activation, suggesting that platelet lipid rafts are dynamic membrane microdomains. Not only actin and fibrin but also small GTPases (Rac, cdc42) and cytoskeleton regulatory proteins (moesin, Arp3, VASP) were detected in the DRM fraction of activated platelets [9]. The possible mechanism of the DRM shift to a higher density in sucrose gradients upon platelet activation presumably involves the high protein-to-lipid ratio [26].
In porcine lung membranes, two distinct types of DRM were obtained after sucrose density gradient centrifugation using Triton X-100. Light DRM contained cerebroside, whereas dense DRM contained Ca2+ATPase and the IP3 receptor [27]. In the adult mouse cerebellum, two distinct types of DRM were also obtained after sucrose density gradient centrifugation using Triton X-100. Light DRM contained cerebroside and sulfatide [28]. In B-lymphocytes, two distinct types of DRM were obtained after sucrose density gradient centrifugation using Brij 98. Light DRM contained ganglioside GM1 and MHC II, whereas dense DRM contained ganglioside GM2 and MHC I [29]. These results suggest endocytosis of MHC molecules by distinct lipid rafts. In HEK293T cells, two distinct types of DRM were also obtained after sucrose density gradient centrifugation using sodium carbonate (pH 11). Light DRM contained ganglioside GM1, whereas dense DRM contained cholesterol and flotillins [30]. Therefore, the platelet DRM shifts in sucrose gradients might be due to changes in lipid composition. Lactosylceramide and ganglioside GM3 are the major glycosphingolipids of human platelets [31]. Resting platelets do not express ganglioside GD3. The stimulation of platelets with ADP resulted in the formation of ganglioside GD3 by GD3 synthesis from the GM3 pool [31][32][31,32]. The GD3 synthase is CMP-NeuAc:NeuAc α2-3Gal β1-4Glc β1-1′Cer α2,8-sialyltransferase [33]. The stimulation of platelets with thrombin showed an increase in the amount of ganglioside GM3 [34]. The stimulation of platelets with ADP showed a decrease in the amount of cholesterol in the DRM raft fraction [10]. The precise mechanism of DRM shifts to a higher density in sucrose gradients upon platelet activation remains to be elucidated.
4. Platelet Glycosphingolipids
- Platelet Glycosphingolipids
Lactosylceramide is the most abundant neutral glycosphingolipid. Its major fatty acids are 20:0, 22:0, 24:0, and 24:1. Ganglioside GM3 is the most abundant acidic glycosphingolipid. The neuraminic acid component was N-acetylneuraminic acid [35][36][35,36]. In addition, galactosylceramide [36], sulfatide [37], glucosylceramide [38][34,38], ganglioside GM1 [39], globotriaosylceramide Gb3 [40], and sialyl-galactosylgloboside [41] are also found in human platelets.
Sulfatide is present on platelet surfaces that bind to adhesive proteins such as vWF, P-selectin, laminin, and thrombospondin [42][43][42,43]. Sulfatide is localized as a large cluster towards the center of spreading platelets [44], suggesting that sulfatide-rich rafts may be platforms involved in intracellular signaling. Sulfatide micelles, the sulfatide-binding recombinant malaria circumsporozoite protein (MCSP), and the sulfatide-specific single-chain fragment variable antibody probe PA38 inhibit this adhesion [44][45][46][44–46]. The sulfatide antagonist MCSP reverses platelet aggregation induced by ADP, collagen, or TRAP [45]. Sulfatide-deficient mice display an extended lag phase of collagen-induced platelet aggregation [44].
The adaptor protein Disabled-2 (Dab2) as a key regulator of platelet signaling is a sulfatide-binding protein. Its interaction is mediated by two N-terminal conserved basic motifs (amino acid residues 24–29 and 49–54) with a dissociation constant Kd of 0.6 μM [47]. Dab2 is present in the cytoplasm and α-granules of platelets and is released from the platelets in response to platelet activation. Dab2 interacts with the cytoplasmic tail of the integrin αIIbβ3 and regulates inside-out signaling [48]. On the other hand, Dab2 released from α-granules inhibit platelet aggregation by competing with fibrinogen for binding to the integrin αIIbβ3, an interaction that is modulated by Dab2 binding to sulfatide at the outer leaflet of the plasma membrane. The Dab2 sulfatide-binding motif peptide can prevent sulfatide-induced platelet aggregation [49][50][49,50]. The bleeding time is prolonged and thrombus formation is impaired in Dab2-deficient mice. Dab2-deficient platelets elicited a selective defect in platelet aggregation and spreading on fibrinogen by thrombin stimulation [51].
Sulfatide on the platelet surface interacts with a blood coagulation factor, playing a major role in hemostasis. Blood coagulation cascade has two pathways: intrinsic pathway and extrinsic pathway. Coagulation factor XII is a plasma serine protease that initiates the intrinsic pathway of blood coagulation upon contact with anionic surfaces, such as sulfatide on the plasma membrane. Annexins (ANXs) are implicated in the regulation of blood coagulation reactions by binding to sulfatide [52]. ANXA3, ANXA4, and ANXA5 inhibit sulfatide-induced plasma coagulation. ANXA4 inhibits sulfatide-induced autoactivation of Factor XII to Factor XIIa and the conversion of its natural substrate Factor XI to Factor XIa [53].
Ganglioside GD3 is rapidly expressed on the platelet surface following platelet activation and internalized to the cytoskeleton where it transiently associates first with the Src family kinase Lyn then with the Fc receptor gamma chain [32]. The binding of bacterial cells to human platelets contributes to the pathogenesis of infective endocarditis. Platelet binding by Streptococcus mitis strain SF100 is mediated by two surface proteins, PblA and PblB. α2-8-linked sialic acid residues on platelet membrane ganglioside GD3 are the primary targets for PblA/PblB-mediated binding to human platelets. [54].
Globotriaosylceramide Gb3 is a functional receptor of the Shiga toxin [40]. Shiga toxin is the principal virulence factor of enterohemorrhagic Escherichia coli. Thrombocytopenia caused by platelet consumption in thrombi is a primary symptom of hemolytic uremic syndrome associated with Shiga toxin. Shiga toxin1 and its B (binding) subunit bind to platelets, leading to fibrinogen binding and platelet aggregation[55] [55]. The possible existence of glycosphingolipid-specific rafts, such as sulfatide-rich rafts, remains to be explored.