Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Peter Tang and Version 1 by Elisa Caffarelli.
RNA metabolism is central to cellular physiopathology. Almost all the molecular pathways underpinning biological processes are affected by the events governing the RNA life cycle, ranging from transcription to degradation. The deregulation of these processes contributes to the onset and progression of human diseases.
- ALS
- motoneurons
- neurodegeneration
- RNA metabolism
- noncoding RNAs
- microRNAs
- long noncoding RNAs
- circular RNAs
1. Introduction
Amyotrophic lateral sclerosis (ALS) is an aging-related and lethal neurodegenerative disorder characterized by the progressive degeneration of motoneurons (MNs) in the spinal cord (SC), brainstem (BS), and motor cortex (MCx). The consequent motor axonal retraction causes muscle weakness and progressive paralysis as major symptoms. Death usually occurs due to respiratory failure, generally within three to five years of onset. However, population-based studies revealed that ALS involves the central nervous system (CNS) more extensively than previously imagined. In particular, up to 50% of ALS patients develop cognitive and behavioral alterations and about 13% have concomitant frontotemporal dementia (FTD), which led to considering ALS and FTD as the two ends of one clinicopathological spectrum [1,2][1][2].
ALS has been classified into familial ALS (fALS), representing about 10% of cases, and sporadic ALS (sALS). They are indistinguishable from a clinical point of view, except for the onset which is earlier in fALS [3]. The latter can be inherited in an autosomal dominant manner and, more rarely, in an autosomal recessive or X-linked manner [4,5][4][5]. Cases of fALS have been attributed to mutations, mostly missense substitutions, in more than 20 genes. Among these, four genes, namely SOD1 (Cu-Zn superoxide dismutase 1), C9ORF72 (hexanucleotide expansion repeat in chromosome 9 open reading frame 72), TARDBP (transactive response DNA-binding protein 43 kDa), and FUS (fused in sarcoma), account for up to 70% of all cases of fALS [5,6,7][5][6][7]. Individuals who do not have affected relatives are classified as sALS patients.
DNA sequencing analyses carried out in patients with sALS revealed that 1–3% of cases are caused by SOD1 mutations [8] and 5% by intronic expansion in C9ORF72 [9]. Mutations in the other ALS-associated genes, such as TARDBP, coding for TDP-43 protein, FUS, HNRNPA1, SQSTM1, VCP, OPTN, and PFN1, are rare in sALS, whose onset could be contributed to by environmental factors [10].
ALS was initially interpreted as a proteostasis failure [10]. This view was supported by the finding that some mutated RNA-binding proteins (RBPs), such as the components of the ribonucleoprotein (RNP) granules TDP-43 and FUS, are delocalized in the cytoplasm where they form pathological aggregates [11]. This phenomenon is exacerbated by the alterations of the two main pathways of protein clearance, namely the ubiquitin–proteasome system [12] and autophagy [13]. However, the same ALS-associated proteins are regulators of RNA metabolism, leading to a further interpretation of the pathology as an RNA disorder. Interacting with thousands of RNA targets, they affect splicing, transport, stability, and even translation, which means that a disturbance in the function of these proteins may affect RNA metabolism on a broad scale [14]. As an example, cross-linking immunoprecipitation (CLIP)-Seq analysis unveiled more than 39,000 TDP-43-binding sites in the mouse transcriptome [15]. Furthermore, the splicing patterns of 965 messenger RNAs (mRNAs), whose products were mainly involved in synaptic activity, were altered upon reduction of the protein from adult mouse brain, Indicating that TDP-43 is key to normal splicing patterns of several brain-enriched mRNAs [15,16][15][16]. Similarly, alternative splicing of mRNAs was altered in FUS-related ALS, with consequent deregulation of neuronal gene expression and production of thousands of aberrantly processed mRNAs [17]. The fact that these ALS-associated proteins intervene not only in the metabolism of mRNAs, with dramatic consequences on protein products, but also in noncoding RNAs (ncRNAs) with an impact on the biological processes they control, is of growing interest. A clear example is the role played by TDP-43 and FUS in the biosynthesis of microRNAs (miRNAs), small ncRNAs that orchestrate differentiation and developmental programs by pleiotropically regulating gene expression [18,19][18][19].
Based on these considerations, ALS has also been proposed as an RNA-mediated neuropathology, which better reflects the heterogeneity of the disease [10].
2. A Brief History of ALS
ALS is also called Lou Gehrig’s disease in the United States and MN disease in the United Kingdom [10]. The name of the pathology reflects both the degeneration of the upper MNs, whose axons project from the cortex to the BS and lateral SC (lateral sclerosis), and the death of lower MNs, which project from the BS or SC to the muscle, causing its wasting (amyotrophy). It was first described as a specific entity in 1869 by the neurologist Jean-Martin Charchot [20]. In the mid-1900s, Kurland and Mulder, carrying out a study on a case series of 58 patients, reported 10% familial cases [21,22][21][22]. More recently, the combination of population-based epidemiological studies with advanced genetics and the development of new bioinformatics tools and neuroimaging techniques led to considering ALS as a syndrome encompassing a wide clinical and pathological spectrum. These findings prompted further stratification of ALS into subtypes, which will be very helpful for the prediction of prognosis and for the design of specific treatments based on different disease mechanisms.
Different criteria have been used for classifying ALS. The traditional definition of ALS subtypes, based on the involvement of upper or lower MNs, was overtaken by other classifications relying on different parameters. A statistical method was developed that predicts prognosis with more accuracy than do clinical phenotypes. It consisted of applying latent class cluster analysis to a large database including 1467 records of ALS patients. This method provided five phenotypic classes of ALS that strongly predicted survival [23]. Another classification of ALS is based on the site of onset and the involvement of different sets of MNs. Accordingly, four forms can be diagnosed: (i) progressive muscular atrophy, which mainly affects spinal neurons or lower MNs and causes limb weakness and atrophy; (ii) primary lateral sclerosis, which primarily affects corticospinal MNs and causes spasticity with increased limb tone; (iii) bulbar ALS, a devastating variant, that mainly affects BS MNs innervating tongue muscles, causing difficulties in speech, chewing, and swallowing; (iv) pseudobulbar palsy, that affects cortical frontobulbar MNs and causes emotion accentuation, absence of facial expression, spastic dysarthria, and dysphagia [10,24][10][24]. To date, none of the used classifications include the cognitive and behavioral symptoms. A range of subtypes should also be highlighted to overcome the heterogeneity of ALS and define subcohorts of patients to address personalized treatments.
3. Face with ALS: Onset, Clinical Manifestation, and Diagnosis
As an aging-related neurodegenerative disease, the occurrence of ALS is growing with the increasing aging of the population [14]. It is the most common adult-onset MN disease diagnosed in 1–2 cases per 100,000 individuals every year in most countries and it is, therefore, considered an orphan disease. However, its inevitably lethal outcome gives incommensurate importance to its occurrence. In the United Kingdom and the United States, ALS determines more than 1 in every 500 deaths in adults, which has led to the prediction that more than 15 million people presently alive across the world will die of the disease [14]. In more detail, population-based studies highlighted that ALS is more common in men than in women [25,26][25][26] and that its incidence differs depending on ancestral origin. It is particularly low in the population of mixed ancestral origin in North America (0.63 cases per 100,000 individuals) [27], whereas it is higher in regions with relatively homogeneous populations, such as in European populations (2.6 cases per 100,000 individuals) [28,29][28][29].
The age of onset is highly variable but almost always occurs in the fifth or sixth decade of life, at a mean age of 55 years. Presumably, it might begin early in the first two decades of life without clear symptoms and emerge only later during life. Median survival is 2 to 4 years from the onset with only 5–10% of patients surviving longer [30,31][30][31]. In particular, many of the long-term survivors show either upper MN or lower MN involvement [32,33][32][33].
Disease onset begins focally and eventually spreads to other body districts. Patients initially experience muscle weakness, fasciculations, muscle atrophy, spasticity, and hyperreflexia that ultimately lead to paralysis [10]. Astrogliosis and microgliosis, accompanied by mitochondrial dysfunction and defects in axonal transport, are hallmarks of the disorder [10].
The diagnosis of ALS is made difficult by the heterogeneous clinical presentation and the absence of a specific test. It relies on a detailed description of the symptoms, physical examination, electrodiagnostic testing, neuroimaging, and familiar history. The El Escorial or Awaji diagnostic criteria are exploited when there is a history of progressive weakness in one or more body regions and evidence of involvement of upper and lower MNs [34]. Thus far, ALS standard treatment consists of multidisciplinary care, including respiratory support and symptom management, whereas the only U.S. Food and Drug Administration-approved drugs are riluzole and edaverone that have only limited effects on patient survival [35].
The absence of effective treatments for the disease is due to the lack of deeper knowledge of the pathogenic mechanisms responsible for MN death, and to the delayed diagnosis usually made in an advanced pathological state. This could be overcome with the identification of reliable biomarkers for early diagnosis, patient stratification, and for the effectiveness of pharmacological therapies.
Many studies are going in this direction. They mainly focus on neurofilaments (Nfs), neuron-specific cytoskeletal proteins that are involved in the stabilization and polarization of neural cells and, therefore, in effective axonal conduction. Notably, their concentration increases in biological fluids proportionally to the degree of axonal damage [36].
Although not yet adopted into clinical practice, the levels of phosphorylated neurofilament heavy chain (pNfH) in cerebrospinal fluid (CSF) have been proposed as specific biomarkers for MN disease. pNfH is endowed with the best performance to discriminate between patients with ALS and healthy and neurological controls with a sensitivity of about 90% [37]. Another study explored blood as an alternative source for measuring pNfH levels. ALS patients displayed elevated concentrations of serum pNfH, that correlated with the disease progression rate [38]. However, given the proximity to the degenerating MNs in the brain and SC, CSF pNfH outperformed serum pNfH (10-fold higher than blood) in discriminating ALS patients [36,39][36][39]. Recently, single-molecule assays allowed the evaluation of ultralow concentrations of blood Nf, which may be very advantageous since blood samples are easily accessible and attainable in a less invasive way compared to CSF [36].
4. RNA Biology of ALS
The protein-coding genes associated with ALS pathogenesis have been grouped into three main classes: the genes altering proteostasis and protein quality control, those involved in cytoskeletal dynamics, and genes affecting RNA metabolism [10]. Recently, great emphasis has been given to the latter gene class and deregulation of RNA has emerged as a major contributor to ALS.
Accordingly, the major ALS-causative genes, namely SOD1, C9ORF72, TARDBP, and FUS, are involved in the control of RNA metabolism to different degrees. In particular, SOD1 negatively affects the stability and function of some mRNA species by interacting with their 3′-untranslated region (3′-UTR) [40,41,42][40][41][42]. The interaction of mutant SOD1 with vascular endothelial growth factor (VEGF) mRNA, besides causing the recruitment of other proteins such as TIAR and HuR into insoluble aggregates, also determines a decrease in VEGF mRNA levels. Similarly, as observed in human spinal MN from SOD1-ALS cases, the binding of mutant SOD1 to neurofilament light chain (NFL) mRNA destabilizes the transcript [40]. The reduction of NFL mRNA levels results in an aberrant stoichiometry of NF subunits, NF aggregation, and neurite degeneration in the iPSC-derived model of ALS [42]. Additionally, mutant SOD1 has been shown to induce alternative splicing deregulation [43].
The C9ORF72 gene could cause ALS through an RNA toxicity mechanism. It carries repeat expansion mutations and accounts for about 50% of fALS and 10% of sALS cases [44]. Both strands of C9ORF72 repeat expansion are transcribed, producing RNA foci that accumulate in patient tissues [45]. The aberrant RNA foci may, in turn, act as a platform that sequesters several RBPs, such as hnRNP-A3, FUS, and TDP-43, producing alterations in RNA metabolism at a global level [46,47,48][46][47][48]. Accordingly, the use of antisense oligonucleotides (ASOs) targeting C9ORF72 repeat expansion avoids RNA foci formation and restore the alteration of gene expression in ALS MNs [46,49][46][49].
Mutations in the TARDBP gene are found in most cases of ALS [50]. Importantly, independent studies carried out in zebrafish [51], Drosophila [52[52][53],53], cultured mammalian neuronal cells [54[54][55][56],55,56], and mice [57] pointed to the relevance of TDP-43 activity as an RNA processing regulator of neuronal differentiation, synaptic transmission, and neuronal plasticity. Several studies underscored its involvement in every step of RNA metabolism [58] as well as its relevant role in miRNA biosynthesis [55,59,60,61,62][55][59][60][61][62].
Mutations in TDP-43 mainly occur in the C terminus, containing the nuclear localization signal, and are responsible for mislocalization of the nuclear protein in the cytoplasm of MNs, where it forms insoluble aggregates. This may cause, at the same time, a loss of function of TDP-43 in the nucleus and a gain of cytoplasmic toxic function, both being detrimental to neuronal function and survival.
As for TDP-43, FUS is a ubiquitously expressed RBP regulating several aspects of RNA metabolism and processing. It is a predominantly nuclear protein crucially involved in transcription, pre-mRNA splicing, and miRNA biogenesis [63]. However, it shuttles to the cytoplasm [64], particularly in neurons, indicating that it may participate in regulating mRNA transport into neurites and local protein translation at synapses [65,66][65][66]. Mutant FUS displays an abnormal cytoplasmic localization in the neurons of ALS patients where it accumulates in cytoplasmic inclusions, the stress granules (SGs) [67,68][67][68].
Interestingly, it was demonstrated that the RNA-binding domain of both TDP-43 and FUS is essential for the neurodegenerative phenotype [69]. In particular, it was shown that RNA-binding-incompetent FUS, also carrying ALS-causing mutations, predominantly localizes in the nucleus in both Drosophila MNs and in a neuronal cell line [69]. This finding reveals that the aberrant cytoplasmic localization of FUS is mediated by its RNA-binding ability, conferring to RNA molecules a relevant role in FUS-ALS pathogenesis [69].
Although much emphasis has been placed on the influence that these ALS causative genes exert on the metabolism of protein-coding RNAs, it is time to complete the biological context of the disease by highlighting the contribution of different classes of ncRNAs with regulatory activities.
Notably, a recent transcriptome profiling of both coding and long noncoding RNAs (lncRNAs) in peripheral blood mononuclear cells of unmutated sALS patients [70] versus healthy controls highlighted that the majority of differentially expressed genes belong to the nonprotein-coding class. In particular, among the 380 differentially expressed genes, 293 were lncRNAs (183 upregulated and 110 downregulated genes) whereas 87 were mRNAs (30 downregulated and 57 upregulated) [71]. It is noteworthy that the high levels of altered noncoding transcripts were not observed in other neurodegenerations such as Alzheimer’s and Parkinson’s disease [71], which supports the hypothesis of a major involvement of the transcriptional machinery in ALS.
5. Noncoding RNA Landscape
Upon the completion of the Human Genome Project, it was realized that of the three billion bases of the human genome, only approximately 2% encode proteins, whereas the most conspicuous portion produces a huge number of so-called ncRNAs [72,73,74][72][73][74]. Notably, their denomination refers to what they are not. In fact, with only some exceptions, they are not endowed with a codogenic potential, having only short open reading frames often interrupted by stop codons. NcRNAs are very diversified, they can be of various sizes, short (less than 200 nt) or long (greater than 200 nt), and have different conformations, both linear and circular (Figure 1). The unifying theme for all these RNAs is their function as fine regulators of gene expression, which eventually orchestrate differentiation and developmental programs through the interaction with other biological macromolecules. Moreover, their high enrichment in the nervous system (NS) led to a tremendous interest in decrypting their role in NS development and function.
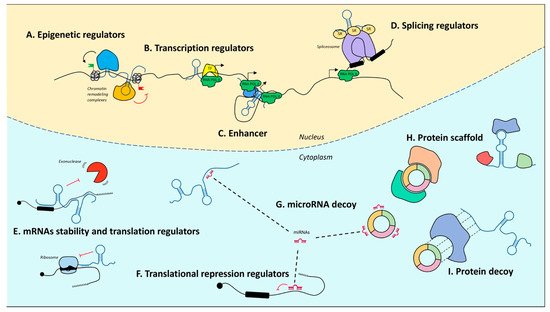
Figure 1. Mechanisms of action of noncoding RNAs. In the nucleus, lncRNAs can regulate gene expression by guiding epigenetic machineries (A), recruiting transcription factors to specific loci (B), acting as enhancers to promote transcription (C), or by recruiting splicing factors (D). In the cytoplasm, lncRNAs modulate mRNA stability and translation (E). MicroRNAs act as translational repressors (F) and may be sponged by both lncRNAs and circRNAs (G). LncRNAs and circRNAs may also act protein scaffolds (H) or decoys (I). T arrows indicate inhibition activity.
References
- Robberecht, W.; Philips, T. The Changing Scene of Amyotrophic Lateral Sclerosis. Nat. Rev. Neurosci. 2013, 14, 248–264.
- Swinnen, B.; Robberecht, W. The Phenotypic Variability of Amyotrophic Lateral Sclerosis. Nat. Rev. Neurol. 2014, 10, 661–670.
- Li, T.M.; Alberman, E.; Swash, M. Comparison of Sporadic and Familial Disease amongst 580 Cases of Motor Neuron Disease. J. Neurol. Neurosurg. Psychiatry 1988, 51, 778–784.
- Pasinelli, P.; Brown, R.H. Molecular Biology of Amyotrophic Lateral Sclerosis: Insights from Genetics. Nat. Rev. Neurosci. 2006, 7, 710–723.
- Andersen, P.M.; Al-Chalabi, A. Clinical Genetics of Amyotrophic Lateral Sclerosis: What Do We Really Know? Nat. Rev. Neurol. 2011, 7, 603–615.
- Chia, R.; Chiò, A.; Traynor, B.J. Novel Genes Associated with Amyotrophic Lateral Sclerosis: Diagnostic and Clinical Implications. Lancet Neurol. 2018, 17, 94–102.
- Chiò, A.; Battistini, S.; Calvo, A.; Caponnetto, C.; Conforti, F.L.; Corbo, M.; Giannini, F.; Mandrioli, J.; Mora, G.; Sabatelli, M.; et al. Genetic Counselling in ALS: Facts, Uncertainties and Clinical Suggestions. J. Neurol. Neurosurg. Psychiatry 2014, 85, 478–485.
- Gamez, J.; Corbera-Bellalta, M.; Nogales, G.; Raguer, N.; García-Arumí, E.; Badia-Canto, M.; Lladó-Carbó, E.; Álvarez-Sabín, J. Mutational Analysis of the Cu/Zn Superoxide Dismutase Gene in a Catalan ALS Population: Should All Sporadic ALS Cases Also Be Screened for SOD1? J. Neurol. Sci. 2006, 247, 21–28.
- Cooper-Knock, J.; Hewitt, C.; Highley, J.R.; Brockington, A.; Milano, A.; Man, S.; Martindale, J.; Hartley, J.; Walsh, T.; Gelsthorpe, C.; et al. Clinico-Pathological Features in Amyotrophic Lateral Sclerosis with Expansions in C9ORF72. Brain 2012, 135, 751–764.
- Taylor, J.P.; Brown, R.H.; Cleveland, D.W. Decoding ALS: From Genes to Mechanism. Nature 2016, 539, 197–206.
- Portz, B.; Lee, B.L.; Shorter, J. FUS and TDP-43 Phases in Health and Disease. Trends Biochem. Sci. 2021, 46, 550–563.
- Deng, H.-X.; Chen, W.; Hong, S.-T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in UBQLN2 Cause Dominant X-Linked Juvenile and Adult-Onset ALS and ALS/Dementia. Nature 2011, 477, 211–215.
- Fecto, F.; Yan, J.; Vemula, S.P.; Liu, E.; Yang, Y.; Chen, W.; Zheng, J.G.; Shi, Y.; Siddique, N.; Arrat, H.; et al. SQSTM1 Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Arch. Neurol. 2011, 68, 1440–1446.
- Wyss-Coray, T. Ageing, Neurodegeneration and Brain Rejuvenation. Nature 2016, 539, 180–186.
- Polymenidou, M.; Lagier-Tourenne, C.; Hutt, K.R.; Huelga, S.C.; Moran, J.; Liang, T.Y.; Ling, S.-C.; Sun, E.; Wancewicz, E.; Mazur, C.; et al. Long Pre-MRNA Depletion and RNA Missplicing Contribute to Neuronal Vulnerability from Loss of TDP-43. Nat. Neurosci. 2011, 14, 459–468.
- Arnold, E.S.; Ling, S.-C.; Huelga, S.C.; Lagier-Tourenne, C.; Polymenidou, M.; Ditsworth, D.; Kordasiewicz, H.B.; McAlonis-Downes, M.; Platoshyn, O.; Parone, P.A.; et al. ALS-Linked TDP-43 Mutations Produce Aberrant RNA Splicing and Adult-Onset Motor Neuron Disease without Aggregation or Loss of Nuclear TDP-43. Proc. Natl. Acad. Sci. USA 2013, 110, E736–E745.
- Butti, Z.; Patten, S.A. RNA Dysregulation in Amyotrophic Lateral Sclerosis. Front. Genet. 2019, 10, 712.
- Gregory, R.I.; Yan, K.; Amuthan, G.; Chendrimada, T.; Doratotaj, B.; Cooch, N.; Shiekhattar, R. The Microprocessor Complex Mediates the Genesis of MicroRNAs. Nature 2004, 432, 235–240.
- Ling, S.-C.; Albuquerque, C.P.; Han, J.S.; Lagier-Tourenne, C.; Tokunaga, S.; Zhou, H.; Cleveland, D.W. ALS-Associated Mutations in TDP-43 Increase Its Stability and Promote TDP-43 Complexes with FUS/TLS. Proc. Natl. Acad. Sci. USA 2010, 107, 13318–13323.
- Charcot, J.-M.; Joffroy, A. Deux Cas d’atrophie Musculaire Progressive Avec Lésions de La Substance Grise et Des Faisceaux Antérolatéraux de La Moelle Épinière. Arch. Physiol. Norm. Pathol. 1869, 2, 744–760.
- Kurland, L.K.; Mulder, D.W. Epidemiologic Investigations of Amyotrophic Lateral Sclerosis: 1. Preliminary Report on Geographic Distribution, with Special Reference to the Mariana Islands, Including Clinical and Pathologic Observations. Neurology 1954, 4, 355–378.
- Mulder, D.W. Epidemiologic Investigations of Amyotrophic Lateral Sclerosis: 2. Familial Aggregations Indicative of Dominant Inheritance Part II. Neurology 1955, 5, 249–268.
- Ganesalingam, J.; Stahl, D.; Wijesekera, L.; Galtrey, C.; Shaw, C.E.; Leigh, P.N.; Al-Chalabi, A. Latent Cluster Analysis of ALS Phenotypes Identifies Prognostically Differing Groups. PLoS ONE 2009, 4, e7107.
- Finegan, E.; Chipika, R.H.; Li Hi Shing, S.; Hardiman, O.; Bede, P. Pathological Crying and Laughing in Motor Neuron Disease: Pathobiology, Screening, Intervention. Front. Neurol. 2019, 10, 260.
- Pape, J.A.; Grose, J.H. The Effects of Diet and Sex in Amyotrophic Lateral Sclerosis. Rev. Neurol. (Paris) 2020, 176, 301–315.
- Trojsi, F.; D’Alvano, G.; Bonavita, S.; Tedeschi, G. Genetics and Sex in the Pathogenesis of Amyotrophic Lateral Sclerosis (ALS): Is There a Link? Int. J. Mol. Sci. 2020, 21, 3647.
- Gordon, P.H.; Mehal, J.M.; Holman, R.C.; Rowland, L.P.; Rowland, A.S.; Cheek, J.E. Incidence of Amyotrophic Lateral Sclerosis Among American Indians and Alaska Natives. JAMA Neurol. 2013, 70, 476–480.
- Logroscino, G.; Traynor, B.J.; Hardiman, O.; Chiò, A.; Mitchell, D.; Swingler, R.J.; Millul, A.; Benn, E.; Beghi, E.; EURALS. Incidence of Amyotrophic Lateral Sclerosis in Europe. J. Neurol. Neurosurg. Psychiatry 2010, 81, 385–390.
- Chiò, A.; Logroscino, G.; Traynor, B.J.; Collins, J.; Simeone, J.C.; Goldstein, L.A.; White, L.A. Global Epidemiology of Amyotrophic Lateral Sclerosis: A Systematic Review of the Published Literature. Neuroepidemiology 2013, 41, 118–130.
- Chiò, A.; Logroscino, G.; Hardiman, O.; Swingler, R.; Mitchell, D.; Beghi, E.; Traynor, B.G.; Eurals Consortium. Prognostic Factors in ALS: A Critical Review. Amyotroph. Lateral Scler. 2009, 10, 310–323.
- del Aguila, M.A.; Longstreth, W.T.; McGuire, V.; Koepsell, T.D.; van Belle, G. Prognosis in Amyotrophic Lateral Sclerosis. Neurology 2003, 60, 813–819.
- Turner, M.R.; Parton, M.J.; Shaw, C.E.; Leigh, P.N.; Al-Chalabi, A. Prolonged Survival in Motor Neuron Disease: A Descriptive Study of the King’s Database 1990–2002. J. Neurol. Neurosurg. Psychiatry 2003, 74, 995–997.
- Chiò, A.; Calvo, A.; Moglia, C.; Mazzini, L.; Mora, G.; PARALS Study Group. Phenotypic Heterogeneity of Amyotrophic Lateral Sclerosis: A Population Based Study. J. Neurol. Neurosurg. Psychiatry 2011, 82, 740–746.
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L. El Escorial Revisited: Revised Criteria for the Diagnosis of Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2009, 1, 293–299.
- Bensimon, G.; Lacomblez, L.; Meininger, V.; ALS/Riulzole Study Group. A Controlled Trial of Riluzole in Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2010, 330, 585–591.
- Gagliardi, D.; Meneri, M.; Saccomanno, D.; Bresolin, N.; Comi, G.P.; Corti, S. Diagnostic and Prognostic Role of Blood and Cerebrospinal Fluid and Blood Neurofilaments in Amyotrophic Lateral Sclerosis: A Review of the Literature. Int. J. Mol. Sci. 2019, 20, 4152.
- Poesen, K.; De Schaepdryver, M.; Stubendorff, B.; Gille, B.; Muckova, P.; Wendler, S.; Prell, T.; Ringer, T.M.; Rhode, H.; Stevens, O.; et al. Neurofilament Markers for ALS Correlate with Extent of Upper and Lower Motor Neuron Disease. Neurology 2017, 88, 2302–2309.
- Falzone, Y.M.; Domi, T.; Agosta, F.; Pozzi, L.; Schito, P.; Fazio, R.; Del Carro, U.; Barbieri, A.; Comola, M.; Leocani, L.; et al. Serum Phosphorylated Neurofilament Heavy-Chain Levels Reflect Phenotypic Heterogeneity and Are an Independent Predictor of Survival in Motor Neuron Disease. J. Neurol. 2020, 267, 1.
- De Schaepdryver, M.; Jeromin, A.; Gille, B.; Claeys, K.G.; Herbst, V.; Brix, B.; Van Damme, P.; Poesen, K. Comparison of Elevated Phosphorylated Neurofilament Heavy Chains in Serum and Cerebrospinal Fluid of Patients with Amyotrophic Lateral Sclerosis. J. Neurol. Neurosurg. Psychiatry 2018, 89, 367–373.
- Menzies, F.M.; Grierson, A.J.; Cookson, M.R.; Heath, P.R.; Tomkins, J.; Figlewicz, D.A.; Ince, P.G.; Shaw, P.J. Selective Loss of Neurofilament Expression in Cu/Zn Superoxide Dismutase (SOD1) Linked Amyotrophic Lateral Sclerosis. J. Neurochem. 2002, 82, 1118–1128.
- Lu, L.; Zheng, L.; Viera, L.; Suswam, E.; Li, Y.; Li, X.; Estévez, A.G.; King, P.H. Mutant Cu/Zn-Superoxide Dismutase Associated with Amyotrophic Lateral Sclerosis Destabilizes Vascular Endothelial Growth Factor MRNA and Downregulates Its Expression. J. Neurosci. 2007, 27, 7929–7938.
- Chen, H.; Qian, K.; Du, Z.; Cao, J.; Petersen, A.; Liu, H.; Blackbourn, L.W.; Huang, C.L.; Errigo, A.; Yin, Y.; et al. Modeling ALS with IPSCs Reveals That Mutant SOD1 Misregulates Neurofilament Balance in Motor Neurons. Cell Stem Cell 2014, 14, 796–809.
- Strong, M.J. The Evidence for Altered RNA Metabolism in Amyotrophic Lateral Sclerosis (ALS). J. Neurol. Sci. 2010, 288, 1–12.
- Majounie, E.; Renton, A.E.; Mok, K.; Dopper, E.G.P.; Waite, A.; Rollinson, S.; Chiò, A.; Restagno, G.; Nicolaou, N.; Simon-Sanchez, J.; et al. Frequency of the C9orf72 Hexanucleotide Repeat Expansion in Patients with Amyotrophic Lateral Sclerosis and Frontotemporal Dementia: A Cross-Sectional Study. Lancet Neurol. 2012, 11, 323–330.
- Gendron, T.F.; Bieniek, K.F.; Zhang, Y.J.; Jansen-West, K.; Ash, P.E.A.; Caulfield, T.; Daughrity, L.; Dunmore, J.H.; Castanedes-Casey, M.; Chew, J.; et al. Antisense Transcripts of the Expanded C9ORF72 Hexanucleotide Repeat Form Nuclear RNA Foci and Undergo Repeat-Associated Non-ATG Translation in C9FTD/ALS. Acta Neuropathol. 2013, 126, 829–844.
- Donnelly, C.J.; Zhang, P.W.; Pham, J.T.; Heusler, A.R.; Mistry, N.A.; Vidensky, S.; Daley, E.L.; Poth, E.M.; Hoover, B.; Fines, D.M.; et al. RNA Toxicity from the ALS/FTD C9ORF72 Expansion Is Mitigated by Antisense Intervention. Neuron 2013, 80, 415–428.
- Lee, Y.B.; Chen, H.J.; Peres, J.N.; Gomez-Deza, J.; Attig, J.; Štalekar, M.; Troakes, C.; Nishimura, A.L.; Scotter, E.L.; Vance, C.; et al. Hexanucleotide Repeats in ALS/FTD Form Length-Dependent RNA Foci, Sequester RNA Binding Proteins, and Are Neurotoxic. Cell Rep. 2013, 5, 1178–1186.
- Mori, K.; Weng, S.-M.; Arzberger, T.; May, S.; Rentzsch, K.; Kremmer, E.; Schmid, B.; Kretzschmar, H.A.; Cruts, M.; Van Broeckhoven, C.; et al. The C9orf72 GGGGCC Repeat Is Translated into Aggregating Dipeptide-Repeat Proteins in FTLD/ALS. Science 2013, 339, 1335–1338.
- Lagier-Tourenne, C.; Baughn, M.; Rigo, F.; Sun, S.; Liu, P.; Li, H.-R.; Jiang, J.; Watt, A.T.; Chun, S.; Katz, M.; et al. Targeted Degradation of Sense and Antisense C9orf72 RNA Foci as Therapy for ALS and Frontotemporal Degeneration. Proc. Natl. Acad. Sci. USA 2013, 110, E4530–E4539.
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133.
- Armstrong, G.A.B.; Drapeau, P. Calcium Channel Agonists Protect against Neuromuscular Dysfunction in a Genetic Model of TDP-43 Mutation in ALS. J. Neurosci. 2013, 33, 1741–1752.
- Lu, L.; Wang, S.; Zheng, L.; Li, X.; Suswam, E.A.; Zhang, X.; Wheeler, C.G.; Nabors, L.B.; Filippova, N.; King, P.H. Amyotrophic Lateral Sclerosis-Linked Mutant SOD1 Sequesters Hu Antigen R (HuR) and TIA-1-Related Protein (TIAR). J. Biol. Chem. 2009, 284, 33989–33998.
- Feiguin, F.; Godena, V.K.; Romano, G.; D’Ambrogio, A.; Klima, R.; Baralle, F.E. Depletion of TDP-43 Affects Drosophila Motoneurons Terminal Synapsis and Locomotive Behavior. FEBS Lett. 2009, 583, 1586–1592.
- Sephton, C.F.; Cenik, C.; Kucukural, A.; Dammer, E.B.; Cenik, B.; Han, Y.H.; Dewey, C.M.; Roth, F.P.; Herz, J.; Peng, J.; et al. Identification of Neuronal RNA Targets of TDP-43-Containing Ribonucleoprotein Complexes. J. Biol. Chem. 2011, 286, 1204–1215.
- Di Carlo, V.; Grossi, E.; Laneve, P.; Morlando, M.; Dini Modigliani, S.; Ballarino, M.; Bozzoni, I.; Caffarelli, E. TDP-43 Regulates the Microprocessor Complex Activity during in Vitro Neuronal Differentiation. Mol. Neurobiol. 2013, 48, 952–963.
- Herzog, J.J.; Deshpande, M.; Shapiro, L.; Rodal, A.A.; Paradis, S. TDP-43 Misexpression Causes Defects in Dendritic Growth. Sci. Rep. 2017, 7, 15656.
- Handley, E.E.; Pitman, K.A.; Dawkins, E.; Young, K.M.; Clark, R.M.; Jiang, T.C.; Turner, B.J.; Dickson, T.C.; Blizzard, C.A. Synapse Dysfunction of Layer V Pyramidal Neurons Precedes Neurodegeneration in a Mouse Model of TDP-43 Proteinopathies. Cereb. Cortex 2017, 27, 3630–3647.
- Ratti, A.; Buratti, E. Physiological Functions and Pathobiology of TDP-43 and FUS/TLS Proteins. J. Neurochem. 2016, 138, 95–111.
- Buratti, E.; De Conti, L.; Stuani, C.; Romano, M.; Baralle, M.; Baralle, F. Nuclear Factor TDP-43 Can Affect Selected MicroRNA Levels. FEBS J. 2010, 277, 2268–2281.
- Kawahara, Y.; Mieda-Sato, A. TDP-43 Promotes MicroRNA Biogenesis as a Component of the Drosha and Dicer Complexes. Proc. Natl. Acad. Sci. USA 2012, 109, 3347–3352.
- Zhang, Z.; Almeida, S.; Lu, Y.; Nishimura, A.L.; Peng, L.; Sun, D.; Wu, B.; Karydas, A.M.; Tartaglia, M.C.; Fong, J.C.; et al. Downregulation of MicroRNA-9 in IPSC-Derived Neurons of FTD/ALS Patients with TDP-43 Mutations. PLoS ONE 2013, 8, e76055.
- King, I.N.; Yartseva, V.; Salas, D.; Kumar, A.; Heidersbach, A.; Ando, D.M.; Stallings, N.R.; Elliott, J.L.; Srivastava, D.; Ivey, K.N. The RNA-Binding Protein TDP-43 Selectively Disrupts MicroRNA-1/206 Incorporation into the RNA-Induced Silencing Complex. J. Biol. Chem. 2014, 289, 14263–14271.
- Colombrita, C.; Onesto, E.; Megiorni, F.; Pizzuti, A.; Baralle, F.E.; Buratti, E.; Silani, V.; Ratti, A. TDP-43 and FUS RNA-Binding Proteins Bind Distinct Sets of Cytoplasmic Messenger RNAs and Differently Regulate Their Post-Transcriptional Fate in Motoneuron-like Cells. J. Biol. Chem. 2012, 287, 15635.
- Zinszner, H.; Sok, J.; Immanuel, D.; Yin, Y.; Ron, D. TLS (FUS) Binds RNA in Vivo and Engages in Nucleo-Cytoplasmic Shuttling. J. Cell Sci. 1997, 110, 1741–1750.
- Fujii, R.; Okabe, S.; Urushido, T.; Inoue, K.; Yoshimura, A.; Tachibana, T.; Nishikawa, T.; Hicks, G.G.; Takumi, T. The RNA Binding Protein TLS Is Translocated to Dendritic Spines by MGluR5 Activation and Regulates Spine Morphology. Curr. Biol. 2005, 15, 587–593.
- Fujii, R.; Takumi, T. TLS Facilitates Transport of MRNA Encoding an Actin-Stabilizing Protein to Dendritic Spines. J. Cell Sci. 2005, 118, 5755–5765.
- Kwiatkowski, T.J.; Bosco, D.A.; LeClerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS Gene on Chromosome 16 Cause Familial Amyotrophic Lateral Sclerosis. Science 2009, 323, 1205–1208.
- Dormann, D.; Haass, C. Fused in Sarcoma (FUS): An Oncogene Goes Awry in Neurodegeneration. Mol. Cell. Neurosci. 2013, 56, 475–486.
- Daigle, G.G.; Lanson, N.A.; Smith, R.B.; Casci, I.; Maltare, A.; Monaghan, J.; Nichols, C.D.; Kryndushkin, D.; Shewmaker, F.; Pandey, U.B. Rna-Binding Ability of FUS Regulates Neurodegeneration, Cytoplasmic Mislocalization and Incorporation into Stress Granules Associated with FUS Carrying ALS-Linked Mutations. Hum. Mol. Genet. 2013, 22, 1193–1205.
- Gagliardi, S.; Zucca, S.; Pandini, C.; Diamanti, L.; Bordoni, M.; Sproviero, D.; Arigoni, M.; Olivero, M.; Pansarasa, O.; Ceroni, M.; et al. Long Non-Coding and Coding RNAs Characterization in Peripheral Blood Mononuclear Cells and Spinal Cord from Amyotrophic Lateral Sclerosis Patients. Sci. Rep. 2018, 8, 2378.
- Garofalo, M.; Pandini, C.; Bordoni, M.; Pansarasa, O.; Rey, F.; Costa, A.; Minafra, B.; Diamanti, L.; Zucca, S.; Carelli, S.; et al. Alzheimer’s, Parkinson’s Disease and Amyotrophic Lateral Sclerosis Gene Expression Patterns Divergence Reveals Different Grade of RNA Metabolism Involvement. Int. J. Mol. Sci. 2020, 21, 9500.
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; et al. The Transcriptional Landscape of the Mammalian Genome. Science 2005, 309, 1559–1563.
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of Transcription in Human Cells. Nature 2012, 489, 101–108.
- Dunham, I.; Kundaje, A.; Aldred, S.F.; Collins, P.J.; Davis, C.A.; Doyle, F.; Epstein, C.B.; Frietze, S.; Harrow, J.; Kaul, R.; et al. An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature 2012, 489, 57–74.
More