Biomolecular condensates are membraneless organelles (MLOs) that form dynamic, chemically distinct subcellular compartments organizing macromolecules such as proteins, RNA, and DNA in unicellular prokaryotic bacteria and complex eukaryotic cells. Separated from surrounding environments, MLOs in the nucleoplasm, cytoplasm, and mitochondria assemble by liquid–liquid phase separation (LLPS) into transient, non-static, liquid-like droplets that regulate essential molecular functions. LLPS is primarily controlled by ATP-dependent post-translational modifications (PTMs) that fine-tune the balance between attractive and repulsive charge states and/or binding motifs of proteins. Aberrant phase separation due to the absence of adequate hydrotropic small molecules such as ATP can cause pathological protein aggregation in diseases such as neurodegenerative disorders. Melatonin is a potent antioxidant capable of protecting cardiolipin and membrane lipids raft domains from peroxidation to support ATPase functionality and ion channel activities that may exert a dominant influence over phase separation in biomolecular condensates during condensate coacervation or dissolution processes that are ATP-dependent.
- melatonin
- biomolecular condensate
- neurodegenerative disorder
- liquid–liquid phase separation
- ATP
- lipid raft
- post-translational modification
- m6A
- RNA
Note: The entry will be online only after author check and submit it.
1. Introduction
1. Introduction
2. ATP Regulates Biomolecular Condensates
2.1. Dimerized ATP Synthase/ATPase Require High-Curvature Lipid Domains
2.2.
Translocation of ATP Dimers to Lipid Rafts Are Cellular Responses to Stress and Stimuli
Biomolecular condensates adapt to changing endogenous or exogenous conditions [3] by continuously fine-tuning biochemical reactions, enriching or excluding biomolecules from their environment [7]. The rapid translocation of mitochondrial ATP synthase to lipid rafts may be integral to these adaptive responses because ATP functions not only as a biological hydrotrope [29][89], increasing the solubility of positively charged, intrinsically disordered proteins [90], but may act as a universal and specific regulator of intrinsically disordered regions (IDRs) capable of altering physicochemical properties, conformation dynamics, assembly, and aggregation [44], in addition to providing phosphates as an energy source to fuel post-translational modifications that regulate the fluctuation of biomolecule phase separation during condensate formation [70][89]. Failure to maintain nanoscopic lipid raft domains with appropriate line tension and membrane elasticity [91] to functionally host dimerized ATPase [92], ATP synthase [86] may contribute to aberrant phase separation, resulting in pathogenic protein aggregates in neurodegeneration [11] and cancer [10][12].
The ability of ATP synthase/ATPase to form dimerized rows on the IMM of mitochondria and other membrane surfaces may be highly dependent upon membrane lipid composition [93] and curvature [86]. Uncontrolled, excess oxidative stress can cause lipid peroxidation [94] which induces pathological changes to membrane lipid composition, including alterations of cardiolipin in IMMs [93][95], as well as changes in membrane curvature that prevent optimal dimerization and the subsequent functioning of ATP synthase/ATPase [96][97]. Insufficient or depletion of ATP can directly impact the physical and functional properties of biomolecular condensates [31][32][70][71]. ATP is not only a biological hydrotrope capable of inhibiting protein LLPS and aggregation at high mM concentrations; it has recently been observed to act as a universal and specific regulator of IDRs, altering their physicochemical properties, conformation dynamics, assembly, and aggregation [44].
3. Melatonin Is a Potent Ancient Antioxidant That Protects ATP Levels to Regulate the Formation and Dissolution of MLOs
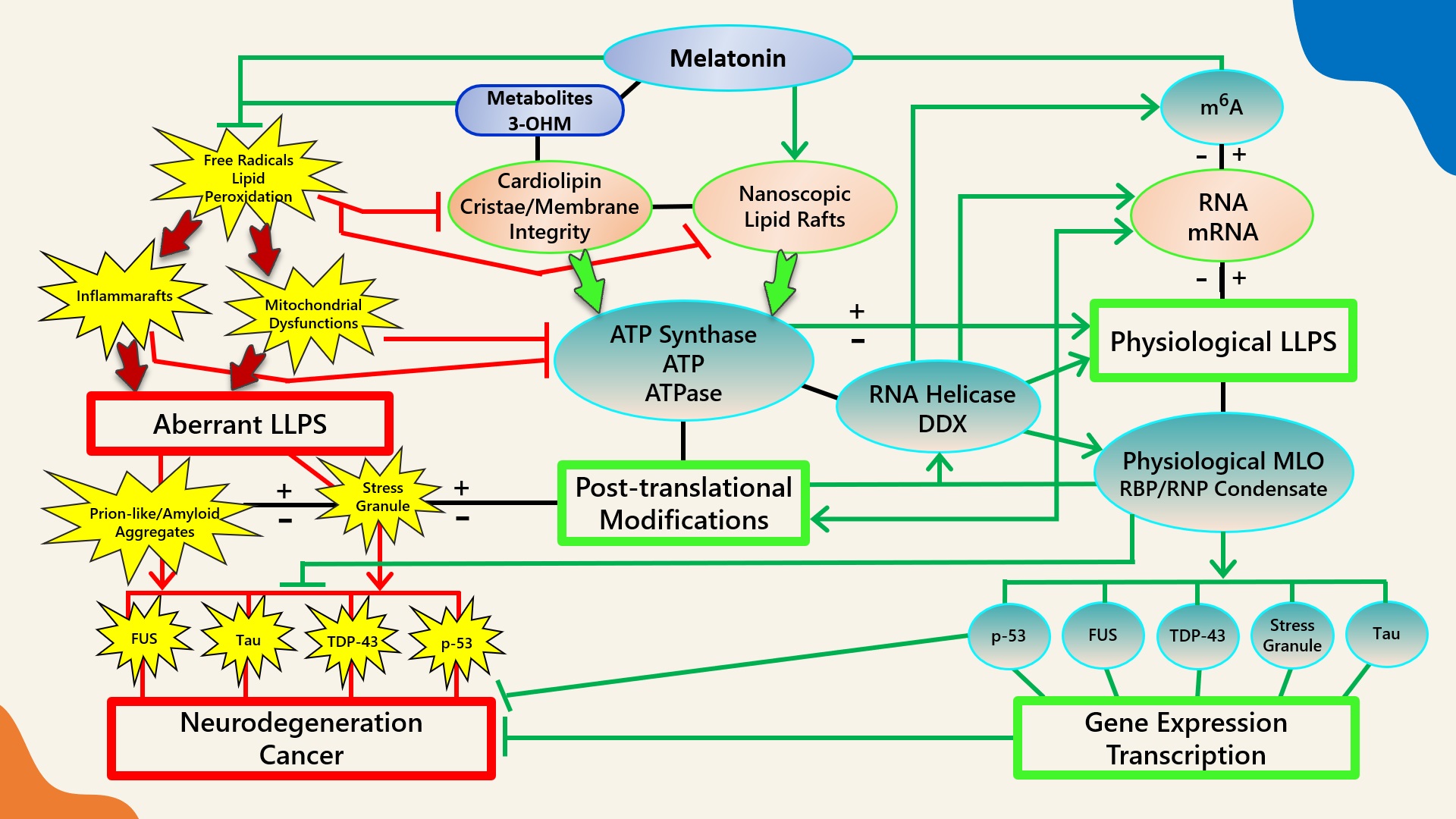 Figure 1. Schematic illustrating the regulation of biomolecular condensates by melatonin represented through observations reported in antioxidant protection against lipid peroxidation to maintain membrane/lipid raft composition/stability that serves to maintain adequate ATP levels in all cellular compartments to fuel, support, and regulate post-translational/m6A modifications that may fine-tune RNA dynamics in the assembly and disassembly of MLOs to prevent pathological aggregations in neurodegenerative disorders. LLPS: liquid–liquid phase separation; DDX: Dead-box RNA helicase; m6A: N6-methyladenosine; MLO: membraneless organelle; RBP: RNA-binding protein; RNP: ribonucleoprotein; PTM: post-translational modification (See Abbreviations for additional acronyms).
Figure 1. Schematic illustrating the regulation of biomolecular condensates by melatonin represented through observations reported in antioxidant protection against lipid peroxidation to maintain membrane/lipid raft composition/stability that serves to maintain adequate ATP levels in all cellular compartments to fuel, support, and regulate post-translational/m6A modifications that may fine-tune RNA dynamics in the assembly and disassembly of MLOs to prevent pathological aggregations in neurodegenerative disorders. LLPS: liquid–liquid phase separation; DDX: Dead-box RNA helicase; m6A: N6-methyladenosine; MLO: membraneless organelle; RBP: RNA-binding protein; RNP: ribonucleoprotein; PTM: post-translational modification (See Abbreviations for additional acronyms).3.1. Melatonin Metabolite 3-OHM Inhibits Lipid Peroxidation by Hydroperoxyl Radical
3.2. Melatonin Is Preferentially Located at Hydrophilic/Hydrophobic Membrane Interfaces
3.3. Melatonin Metabolite Free Radical Scavenging Cascades Rescue Cardiolipin from Hydroperoxyl Radicals (•OOH)
3.4.
Melatonin Antioxidant Cascades May Inhibit NLRP3 Prionoid-Like Aggregation in an ATP-Dependent Manner
Cardiolipin (CL) is a mitochondria signature lipid distinctly attracted to membrane lipid domains with strong negative curvatures, such as the apex of IMM cristae [202][203]. CL is often externalized to the outer mitochondrial membrane (OMM) upon mitochondrial distress from ROS attacks [204][205], whereas oxidized CL in OMM initiates apoptotic signaling processes [206] that can lead to opening of the mitochondrial permeability transition pore (mPTP) and the release of cytochrome c (Cyt c) [207][208]. Externalized CL, whether oxidized or not, becomes an essential signaling platform that binds and interacts with important mitophagic, autophagic, and inflammatory enzymes [205][209], including Beclin 1 [210], tBid, Bax [208][211], caspase-8 [212], and the NLR pyrin domain containing 3 (NLRP3) inflammasomes [213]. A major source of extremely inflammatory cytokines IL-1β and IL-18 [214], NLRP3 inflammasome is a phase-separated supramolecular complex that mediates immune responses upon the detection of cellular stress and dysfunction [215][216][217]. The activation of the NLRP3 inflammasome in macrophages is induced by oxidized phospholipids [218], whereas the docking of externalized CL to NLRP3 inflammasome primes its assembly and subsequent activation in mitochondria [213] as well as mitochondria-associated membranes (MAMs), a region comprising highly specialized proteins which is tethered to the endoplasmic reticulum (ER) [219][220].
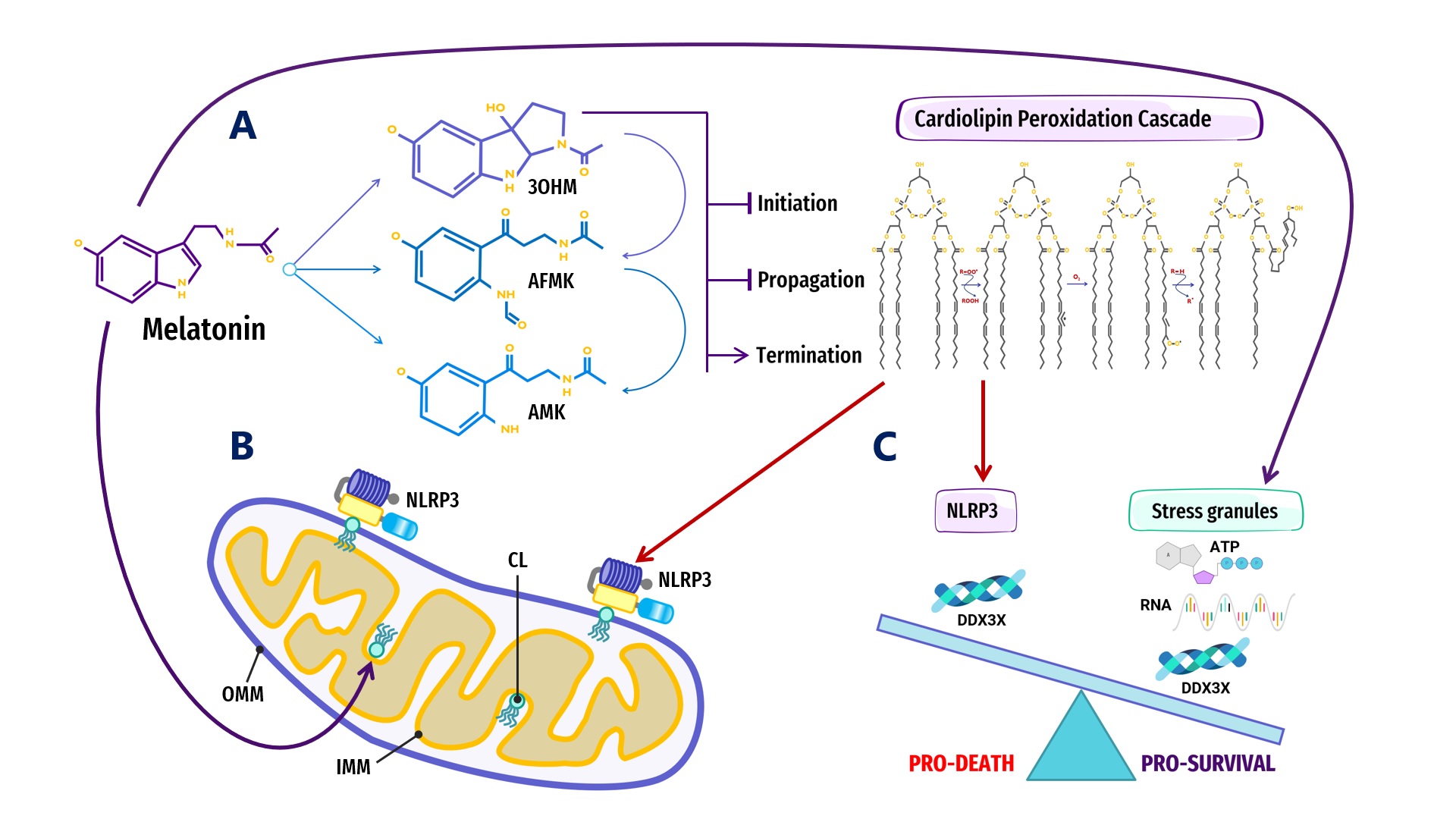 Figure 2. Overview of melatonin regulation of NLRP3 inflammasome (NLRP3) formation, assembly and activation: (A) Summary of melatonin and metabolite antioxidant cascade inhibiting the initiation and propagation of cardiolipin (CL) peroxidation, effectively terminating the CL peroxidation cascade; (B) Oxidized CL is externalized from the cristae/inner mitochondrial membrane (IMM) to the outer mitochondrial membrane (OMM) where it docks and primes NLRP3 inflammasome assembly prior to activation in mitochondria; (C) DDX3X, an ATP-dependent DEAD-box RNA helicase, is the mediator that selects the formation of “Pro-Survival” stress granules or the transition of the NLRP3 inflammasome into “Pro-Death”, stable, prionoid-like complexes. The successful formation of stress granules is also dependent upon the availability of ATP and RNA, both of which may be regulated by melatonin (See Abbreviations for additional acronyms).
Figure 2. Overview of melatonin regulation of NLRP3 inflammasome (NLRP3) formation, assembly and activation: (A) Summary of melatonin and metabolite antioxidant cascade inhibiting the initiation and propagation of cardiolipin (CL) peroxidation, effectively terminating the CL peroxidation cascade; (B) Oxidized CL is externalized from the cristae/inner mitochondrial membrane (IMM) to the outer mitochondrial membrane (OMM) where it docks and primes NLRP3 inflammasome assembly prior to activation in mitochondria; (C) DDX3X, an ATP-dependent DEAD-box RNA helicase, is the mediator that selects the formation of “Pro-Survival” stress granules or the transition of the NLRP3 inflammasome into “Pro-Death”, stable, prionoid-like complexes. The successful formation of stress granules is also dependent upon the availability of ATP and RNA, both of which may be regulated by melatonin (See Abbreviations for additional acronyms).ATP-dependent DEAD-box RNA helicases (DDXs) are ATPases that post-translationally regulate RNA-containing phase-separated organelles in prokaryotes and eukaryotes [227][228]. DDXs promote phase separation in their ATP-bound form, but can also release RNA and induce compartment turnover using ATP hydrolysis. Inhibition of DDX ATPase activity can disrupt the disassembly of physiological MLOs such as P-bodies and stress granules [68][69] (Figure 1). Phosphorylation is one of the most important PTMs that can control the assembly/disassembly of MLOs [229] as well as stabilize or destabilize MLOs including G bodies [230] and p53 [231]. Cells rely on phosphorylation as rapid, reversible responses to different stimuli by changing the physicochemical properties of proteins during phase separation multivalent interactions [70][232]. Phosphorylation establishes covalent bonds between phosphoryl and amino acid hydroxyl groups using the terminal phosphate group in ATP [233]. The ATP-dependent DEAD-box helicase [227] DDX3X responsible for initiating NLRP3 inflammasome aggregation is dependent upon phosphorylation-associated IFN promoter stimulation [224][234][235][236]. When the conserved, eukaryotic, integrated stress response (ISR) pathway is activated by external stress stimuli including hypoxia, nutrient deprivation, viral infections, as well as intrinsic ER stress [237], the phosphorylation of eukaryotic translation initiation factor 2 alpha (eIF2a) on Ser51 [238][239] triggers the formation of stress granules as adaptive homeostatic responses to promote survival and restore homeostasis [240][241][242][243]. It is presently unknown what prompts DDX3X to select the aggregation of pro-survival stress granules over pro-death NLRP3 inflammasomes or vice versa [224][234]. It would not be unreasonable to assume that an excessive oxidative local environment with pathological, enlarged lipid rafts (inflammarafts) [244][245] in membranes could exert a decisive influence over the selection process (Figure 2).
3.5. Melatonin Maintains a High Cytosolic ATP:ADP Ratio through the Optimization of VDAC-CYB5R3 Redox Complexes in Lipid Rafts
Lipid rafts are phase-separated regions in lipid bilayers responsible for important biological functions including signal transduction [271][272] as well as the trafficking and sorting of proteins and lipids [273][274]. The fact that lipid rafts are also important redox signaling platforms that assemble, recruit, and activate redox regulatory multiprotein complex NADPH oxidase [275][276], and host the quintessential plasma membrane redox enzyme complex VDAC-CYB5R3 [277][278], emphasizes the relevance of melatonin as an antioxidant in the protection and stabilization of lipid raft domains.
Nanoscopic transient lipid raft domains in biological membranes are formed by phase separation in response to external stimuli [271][272][279]. Even though cells may alter lipid constituents to control the composition and size of lipid rafts [280], the propagation of molecular stress, lipid raft rattling dynamics and relaxation are some of the basic mechanisms underlying phase separation on the molecular level [281]. The presence of hydrophobic molecules such as melatonin can modulate viscoelastic dynamics through the accumulation and propagation of stress in lipid–lipid interactions [281][282]. Adding melatonin to membrane models led to a breakdown of out-of-phase membrane displacement patterns and the disruption of the vibrational landing platform of lipid biomolecules at the water–membrane interface, effectively slowing the permeation of ROS and other small molecules [281][133].
In 2005, melatonin was first observed to induce phase-separation in DPPC lipid bilayers [250]; recently, melatonin has been observed to modify lipid hydrocarbon chain order to promote phase separation in ternary membrane models [283]. Due to a preference to localize at membrane interfaces [154], melatonin can form strong hydrogen bonds with membrane lipid anionic headgroups that could significantly modulate lipid acyl chain flexibility and lipid dynamics [250]. Melatonin is able to directly interact with cholesterol [284] and displaced cholesterol due to competitive binding to lipid molecules, increasing disorder in the Ld phase to drive cholesterol into the ordered Lo phase [283]. These subtle changes in lipid nanodomains can profoundly affect amyloid processing at membrane sites. Aβ1–40 and Aβ1–42 peptides are known to interact strongly with negatively charged lipids by binding to anionic, negatively charged membranes [285][286][287][288][289]. Increasing cholesterol content lowered the surface charge of lipid membranes in saline solution from positive to negative [290]. Although cholesterol is an indispensable constituent of lipid rafts [271][291], its electrostatic properties altered interactions of charged or polar biomolecules on lipid membrane surfaces and attracted the targeted binding of Aβ deposits at lipid membranes [292][293][294][295].
Local variations in melatonin concentration also affected the re-ordering of lipids in membranes. At 0.5 mol% concentration, melatonin was documented to penetrate lipid bilayers to form fluid domains that enriched lipid membranes where melatonin molecules aligned parallel to phospholipid tails with the electron-dense regions slightly below hydrophilic headgroups; however, at 30 mol% concentration, melatonin molecules aligned parallel to the lipid bilayer, close to the headgroup regions where one melatonin molecule was associated with two lipid molecules to form an ordered, uniform, lateral membrane structure distributed evenly throughout the membrane model [255]. Variations in local concentration and conformational changes in melatonin molecules can directly impact the lipid phase transition, line tension, size, health, and functions of lipid rafts.
Present in all eukaryotes [296], CYB5R3 encodes for a NADH-cytochrome b5 reductase 3 flavoprotein that is engaged in the one-electron transfer from NADH to cytochrome b5 or plasma membrane coenzyme Q, producing NAD+ as a result [297][298]. The soluble isoform of CYB5R3 is exclusive to erythrocytes [299], whereas the membrane-bound isoform is anchored to MOM, ER, and plasma membrane lipid rafts [278][300][301]. Importantly, the OMM-bound CYB5R3 enzyme, ubiquitously expressed in all mammalian cells, is functionally attached to the voltage-dependent anion channel 1 (VDAC1), one of the most prevalent proteins located in the OMM [302][303].
Originally known as mitochondrial porin after its identification in yeast (1985) [304] and humans (1989) [305], VDAC was subsequently observed as a resident protein of lipid rafts in the plasma membranes of animal hearts, brains, and lungs [306] from different human cell lines, including epithelial cells, astrocytes, and neurons [307][308]. Aberrant lipid composition in neuronal lipid rafts disturbs physiological VDAC protein interactions that can affect the opening and closing of VDAC channels, resulting in oxidative stress and neuronal impairments prominent in most AD pathologies [307]. The force-from-lipid principle dictates that the opening and closing of membrane embedded channels can be propelled by the mechanical properties of surrounding lipids [309][310][311][312] and their composition. Changes to raft thickness, curvature and elasticity [313] as a result of lipid peroxidation can therefore affect physiological functions of the VDAC and CYB5R3 redox complex.
CYB5R3 enzymes form large redox centers in lipid rafts that enhance mitochondrial respiration rate and ATP production, albeit resulting in increased production of ROS [278][300][301]. Over stimulation and clustering of CYB5R3 induced oxidative stress-mediated apoptosis of cerebellar granule neurons [314]. Independent of respiratory chain activities, the ascorbate-dependent NADH: cytochrome c oxidoreductase oxidation of NADH at CYB5R3 centers in lipid rafts is also a major source of extracellular superoxide [303][315][316][317][318] that can initiate lipid peroxidation. In Wistar rats, the deregulation of CYB5R3 promptly triggers apoptosis due to the overproduction of superoxide anions at neuronal plasma membranes [278][315]. Excess NADH due to CYB5R3 redox dysfunction can close VDAC, suppressing OXPHOS and increasing glycolysis [303][319], whereas the opening of VDAC also elevates ROS from increased OXPHOS activities [40]. As the most abundant protein in the MOM, VDAC is regarded as a dynamic regulator of mitochondrial functions, interacting with over 100 proteins in health and disease [320]. VDAC opening is believed to globally control mitochondrial metabolism and ROS formation, modulating mitochondria and cellular bioenergetics [40][321]. Nevertheless, the question of whether apoptosis is associated with the opening [322] or closure [323][324] of VDAC has been highly debated [325], further emphasizing the important role of this protein in the regulation of cell life and death [320][326].
VDAC is the gatekeeper which controls the export of ATP out of mitochondria into cytosol and the import of essential respiratory substrates such as ADP and Pi into mitochondria [323][327]; therefore, VDAC opening may be instrumental in determining the fate of MLO formation, regulation, and dissolution. ATP is not only a biological hydrotrope capable of inhibiting protein LLPS and aggregation at high mM concentrations, but it has recently been observed to act as a universal and specific regulator of IDRs capable of altering physicochemical properties, conformation dynamics, assembly, and the aggregation of MLOs [44]. Not only is the preservation of lipid raft structure and composition essential for maintaining specific ion channel properties [307], the amount of cytosolic ATP is dependent upon mitochondrial synthesis and the integrity of CL enriched raft-like lipid domains in mitochondria [277][328][329][330].
3.6. Melatonin May Regulate Glycolytic G Bodies by Increasing ATP
4. Conclusions
The physiological and pathological functions of biomolecular condensates in health and disease may be shaped by powerful, complex, interdependent relationships between membraneless organelles, membranes/lipid rafts, ATP, and most of all, stress and its timely resolution. Melatonin’s intimate association with each of these decisive influencers may position the potent, ancient antioxidant as an important mediator of the phase separation of condensates in health and disease via principal ATP-dependent post-translational mechanisms and regulation of ATP levels in mitochondria and cytoplasm (Figure 1). This novel theoretical review highlights the important connections between melatonin and ATP in the regulation of biomolecular condensates with the intention to spur further research interest and exploration in the full, multi-faceted potential of melatonin that may provide solutions and answers to existing and future challenges and questions in this exciting and promising field of study.
References
- Feng, Z.; Chen, X.; Wu, X.; Zhang, M. Formation of Biological Condensates via Phase Separation: Characteristics, Analytical Methods, and Physiological Implications. J. Biol. Chem. 2019, 294, 14823–14835.
- Oparin, A.I.; Synge, A. The Origin of Life on the Earth/Translated from the Russian by Ann Synge; Elsevier Science Ltd.: Amsterdam, The Netherlands, 1957.
- Riback, J.A.; Katanski, C.D.; Kear-Scott, J.L.; Pilipenko, E.V.; Rojek, A.E.; Sosnick, T.R.; Drummond, D.A. Stress-Triggered Phase Separation is an Adaptive, Evolutionarily Tuned Response. Cell 2017, 168, 1028–1040.e19.
- Li, P.; Banjade, S.; Cheng, H.-C.; Kim, S.; Chen, B.; Guo, L.; Llaguno, M.; Hollingsworth, J.V.; King, D.S.; Banani, S.F.; et al. Phase Transitions in the Assembly of Multivalent Signalling Proteins. Nature 2012, 483, 336–340.
- Su, X.; Ditlev, J.A.; Hui, E.; Xing, W.; Banjade, S.; Okrut, J.; King, D.S.; Taunton, J.; Rosen, M.K.; Vale, R.D. Phase Separation of Signaling Molecules Promotes T Cell Receptor Signal Transduction. Science 2016, 352, 595–599.
- Laflamme, G.; Mekhail, K. Biomolecular Condensates as Arbiters of Biochemical Reactions inside the Nucleus. Commun. Biol. 2020, 3, 773.
- Ditlev, J.A.; Case, L.B.; Rosen, M.K. Who’s In and Who’s Out-Compositional Control of Biomolecular Condensates. J. Mol. Biol. 2018, 430, 4666–4684.
- Shin, Y.; Brangwynne, C.P. Liquid Phase Condensation in Cell Physiology and Disease. Science 2017, 357, eaaf4382.
- Alberti, S.; Dormann, D. Liquid-Liquid Phase Separation in Disease. Annu. Rev. Genet. 2019, 53, 171–194.
- Boija, A.; Klein, I.A.; Young, R.A. Biomolecular Condensates and Cancer. Cancer Cell 2021, 39, 174–192.
- Zbinden, A.; Pérez-Berlanga, M.; De Rossi, P.; Polymenidou, M. Phase Separation and Neurodegenerative Diseases: A Disturbance in the Force. Dev. Cell 2020, 55, 45–68.
- Taniue, K.; Akimitsu, N. Aberrant Phase Separation and Cancer. FEBS J. 2021.
- Hyman, A.A.; Weber, C.A.; Jülicher, F. Liquid-Liquid Phase Separation in Biology. Annu. Rev. Cell Dev. Biol. 2014, 30, 39–58.
- Ahlers, J.; Adams, E.M.; Bader, V.; Pezzotti, S.; Winklhofer, K.F.; Tatzelt, J.; Havenith, M. The Key Role of Solvent in Condensation: Mapping Water in Liquid-Liquid Phase-Separated FUS. Biophys. J. 2021, 120, 1266–1275.
- Lodish, H.; Berk, A.; Lawrence Zipursky, S.; Matsudaira, P.; Baltimore, D.; Darnell, J. Biochemical Energetics; W. H. Freeman: New York, NY, USA, 2000.
- Manchester, K.L. Free Energy ATP Hydrolysis and Phosphorylation Potential. Biochem. Educ. 1980, 8, 70–72.
- Peth, A.; Uchiki, T.; Goldberg, A.L. ATP-Dependent Steps in the Binding of Ubiquitin Conjugates to the 26S Proteasome That Commit to Degradation. Mol. Cell 2010, 40, 671–681.
- Callis, J. The Ubiquitination Machinery of the Ubiquitin System. Arab. Book 2014, 12, e0174.
- Zhao, X. SUMO-Mediated Regulation of Nuclear Functions and Signaling Processes. Mol. Cell 2018, 71, 409–418.
- Van Damme, E.; Laukens, K.; Dang, T.H.; Van Ostade, X. A Manually Curated Network of the PML Nuclear Body Interactome Reveals an Important Role for PML-NBs in SUMOylation Dynamics. Int. J. Biol. Sci. 2010, 6, 51–67.
- Hardenberg, M.; Horvath, A.; Ambrus, V.; Fuxreiter, M.; Vendruscolo, M. Widespread Occurrence of the Droplet State of Proteins in the Human Proteome. Proc. Natl. Acad. Sci. USA 2020, 117, 33254–33262.
- Hondele, M.; Heinrich, S.; De Los Rios, P.; Weis, K. Membraneless Organelles: Phasing out of Equilibrium. Emerg. Top Life Sci. 2020, 4, 331–342.
- Garcia-Jove Navarro, M.; Kashida, S.; Chouaib, R.; Souquere, S.; Pierron, G.; Weil, D.; Gueroui, Z. RNA Is a Critical Element for the Sizing and the Composition of Phase-Separated RNA-Protein Condensates. Nat. Commun. 2019, 10, 3230.
- Elbaum-Garfinkle, S.; Kim, Y.; Szczepaniak, K.; Chen, C.C.-H.; Eckmann, C.R.; Myong, S.; Brangwynne, C.P. The Disordered P Granule Protein LAF-1 Drives Phase Separation into Droplets with Tunable Viscosity and Dynamics. Proc. Natl. Acad. Sci. USA 2015, 112, 7189–7194.
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162, 1066–1077.
- Niaki, A.G.; Sarkar, J.; Cai, X.; Rhine, K.; Vidaurre, V.; Guy, B.; Hurst, M.; Lee, J.C.; Koh, H.R.; Guo, L.; et al. Loss of Dynamic RNA Interaction and Aberrant Phase Separation Induced by Two Distinct Types of ALS/FTD-Linked FUS Mutations. Mol. Cell 2020, 77, 82–94.e4.
- Mann, J.R.; Gleixner, A.M.; Mauna, J.C.; Gomes, E.; DeChellis-Marks, M.R.; Needham, P.G.; Copley, K.E.; Hurtle, B.; Portz, B.; Pyles, N.J.; et al. RNA Binding Antagonizes Neurotoxic Phase Transitions of TDP-43. Neuron 2019, 102, 321–338.e8.
- Wegmann, S.; Eftekharzadeh, B.; Tepper, K.; Zoltowska, K.M.; Bennett, R.E.; Dujardin, S.; Laskowski, P.R.; MacKenzie, D.; Kamath, T.; Commins, C.; et al. Tau Protein Liquid-Liquid Phase Separation Can Initiate Tau Aggregation. EMBO J. 2018, 37, e98049.
- Patel, A.; Malinovska, L.; Saha, S.; Wang, J.; Alberti, S.; Krishnan, Y.; Hyman, A.A. ATP as a Biological Hydrotrope. Science 2017, 356, 753–756.
- Hatzopoulos, M.H.; Eastoe, J.; Dowding, P.J.; Rogers, S.E.; Heenan, R.; Dyer, R. Are Hydrotropes Distinct from Surfactants? Langmuir 2011, 27, 12346–12353.
- Jain, S.; Wheeler, J.R.; Walters, R.W.; Agrawal, A.; Barsic, A.; Parker, R. ATPase-Modulated Stress Granules Contain a Diverse Proteome and Substructure. Cell 2016, 164, 487–498.
- Brangwynne, C.P.; Mitchison, T.J.; Hyman, A.A. Active Liquid-like Behavior of Nucleoli Determines Their Size and Shape in Xenopus Laevis Oocytes. Proc. Natl. Acad. Sci. USA 2011, 108, 4334–4339.
- Pal, S.; Paul, S. ATP Controls the Aggregation of Aβ16-22 Peptides. J. Phys. Chem. B 2020, 124, 210–223.
- Zhang, C.; Rissman, R.A.; Feng, J. Characterization of ATP Alternations in an Alzheimer’s Disease Transgenic Mouse Model. J. Alzheimers. Dis. 2015, 44, 375–378.
- Salis, A.; Ninham, B.W. Models and Mechanisms of Hofmeister Effects in Electrolyte Solutions, and Colloid and Protein Systems Revisited. Chem. Soc. Rev. 2014, 43, 7358–7377.
- Mandl, I.; Grauer, A.; Neuberg, C. Solubilization of Insoluble Matter in Nature; I. The Part Played by Salts of Adenosinetriphosphate. Biochim. Biophys. Acta 1952, 8, 654–663.
- Mehringer, J.; Do, T.-M.; Touraud, D.; Hohenschutz, M.; Khoshsima, A.; Horinek, D.; Kunz, W. Hofmeister versus Neuberg: Is ATP Really a Biological Hydrotrope? Cell Rep. Phys. Science 2021, 2, 100343.
- Schwenke, W.D.; Soboll, S.; Seitz, H.J.; Sies, H. Mitochondrial and Cytosolic ATP/ADP Ratios in Rat Liver in Vivo. Biochem. J 1981, 200, 405–408.
- Imamura, H.; Nhat, K.P.H.; Togawa, H.; Saito, K.; Iino, R.; Kato-Yamada, Y.; Nagai, T.; Noji, H. Visualization of ATP Levels inside Single Living Cells with Fluorescence Resonance Energy Transfer-Based Genetically Encoded Indicators. Proc. Natl. Acad. Sci. USA 2009, 106, 15651–15656.
- Fang, D.; Maldonado, E.N. VDAC Regulation: A Mitochondrial Target to Stop Cell Proliferation. Adv. Cancer Res. 2018, 138, 41–69.
- Ruprecht, J.J.; King, M.S.; Zögg, T.; Aleksandrova, A.A.; Pardon, E.; Crichton, P.G.; Steyaert, J.; Kunji, E.R.S. The Molecular Mechanism of Transport by the Mitochondrial ADP/ATP Carrier. Cell 2019, 176, 435–447.e15.
- Liu, Y.; Chen, X.J. Adenine Nucleotide Translocase, Mitochondrial Stress, and Degenerative Cell Death. Oxid. Med. Cell. Longev. 2013, 2013, 146860.
- Depaoli, M.R.; Karsten, F.; Madreiter-Sokolowski, C.T.; Klec, C.; Gottschalk, B.; Bischof, H.; Eroglu, E.; Waldeck-Weiermair, M.; Simmen, T.; Graier, W.F.; et al. Real-Time Imaging of Mitochondrial ATP Dynamics Reveals the Metabolic Setting of Single Cells. Cell Rep. 2018, 25, 501–512.e3.
- Song, J. Adenosine Triphosphate Energy-Independently Controls Protein Homeostasis with Unique Structure and Diverse Mechanisms. Protein Sci. 2021, 30, 1277–1293.
- Takaine, M.; Imamura, H.; Yoshida, S. High and Stable ATP Levels Prevent Aberrant Intracellular Protein Aggregation. bioRxiv 2021, 2021, 801738.
- Sama, R.R.K.; Ward, C.L.; Kaushansky, L.J.; Lemay, N.; Ishigaki, S.; Urano, F.; Bosco, D.A. FUS/TLS Assembles into Stress Granules and Is a Prosurvival Factor during Hyperosmolar Stress. J. Cell. Physiol. 2013, 228, 2222–2231.
- Mahboubi, H.; Stochaj, U. Cytoplasmic Stress Granules: Dynamic Modulators of Cell Signaling and Disease. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 884–895.
- Hilliker, A. Analysis of RNA Helicases in P-Bodies and Stress Granules. Methods Enzymol. 2012, 511, 323–346.
- Sathyanarayanan, U.; Musa, M.; Bou Dib, P.; Raimundo, N.; Milosevic, I.; Krisko, A. ATP Hydrolysis by Yeast Hsp104 Determines Protein Aggregate Dissolution and Size in Vivo. Nat. Commun. 2020, 11, 5226.
- Kang, J.; Lim, L.; Song, J. ATP Enhances at Low Concentrations but Dissolves at High Concentrations Liquid-Liquid Phase Separation (LLPS) of ALS/FTD-Causing FUS. Biochem. Biophys. Res. Commun. 2018, 504, 545–551.
- Kang, J.; Lim, L.; Song, J. ATP Binds and Inhibits the Neurodegeneration-Associated Fibrillization of the FUS RRM Domain. Commun. Biol. 2019, 2, 223.
- Dang, M.; Lim, L.; Kang, J.; Song, J. ATP Biphasically Modulates LLPS of TDP-43 PLD by Specifically Binding Arginine Residues. Commun. Biol. 2021, 4, 714.
- Heo, C.E.; Han, J.Y.; Lim, S.; Lee, J.; Im, D.; Lee, M.J.; Kim, Y.K.; Kim, H.I. ATP Kinetically Modulates Pathogenic Tau Fibrillations. ACS Chem. Neurosci. 2020, 11, 3144–3152.
- Farid, M.; Corbo, C.P.; Alonso, A.D.C. Tau Binds ATP and Induces Its Aggregation. Microsc. Res. Tech. 2014, 77, 133–137.
- Newby, G.A.; Lindquist, S. Blessings in Disguise: Biological Benefits of Prion-like Mechanisms. Trends Cell Biol. 2013, 23, 251–259.
- Li, L.; McGinnis, J.P.; Si, K. Translational Control by Prion-like Proteins. Trends Cell Biol. 2018, 28, 494–505.
- Schuster, B.S.; Dignon, G.L.; Tang, W.S.; Kelley, F.M.; Ranganath, A.K.; Jahnke, C.N.; Simpkins, A.G.; Regy, R.M.; Hammer, D.A.; Good, M.C.; et al. Identifying Sequence Perturbations to an Intrinsically Disordered Protein That Determine Its Phase-Separation Behavior. Proc. Natl. Acad. Sci. USA 2020, 117, 11421–11431.
- Harmon, T.S.; Holehouse, A.S.; Rosen, M.K.; Pappu, R.V. Intrinsically Disordered Linkers Determine the Interplay between Phase Separation and Gelation in Multivalent Proteins. Elife 2017, 6, e30294.
- Owen, I.; Shewmaker, F. The Role of Post-Translational Modifications in the Phase Transitions of Intrinsically Disordered Proteins. Int. J. Mol. Sci. 2019, 20, 5501.
- Van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of Intrinsically Disordered Regions and Proteins. Chem. Rev. 2014, 114, 6589–6631.
- Küffner, A.M.; Linsenmeier, M.; Grigolato, F.; Prodan, M.; Zuccarini, R.; Capasso Palmiero, U.; Faltova, L.; Arosio, P. Sequestration within Biomolecular Condensates Inhibits Aβ-42 Amyloid Formation. Chem. Sci. 2021, 12, 4373–4382.
- Luo, Y.; Na, Z.; Slavoff, S.A. P-Bodies: Composition, Properties, and Functions. Biochemistry 2018, 57, 2424–2431.
- Stoecklin, G.; Kedersha, N. Relationship of GW/P-Bodies with Stress Granules. Adv. Exp. Med. Biol. 2013, 768, 197–211.
- Kedersha, N.; Stoecklin, G.; Ayodele, M.; Yacono, P.; Lykke-Andersen, J.; Fritzler, M.J.; Scheuner, D.; Kaufman, R.J.; Golan, D.E.; Anderson, P. Stress Granules and Processing Bodies Are Dynamically Linked Sites of mRNP Remodeling. J. Cell Biol. 2005, 169, 871–884.
- Nostramo, R.; Xing, S.; Zhang, B.; Herman, P.K. Insights into the Role of P-Bodies and Stress Granules in Protein Quality Control. Genetics 2019, 213, 251–265.
- Teixeira, D.; Sheth, U.; Valencia-Sanchez, M.A.; Brengues, M.; Parker, R. Processing Bodies Require RNA for Assembly and Contain Nontranslating mRNAs. RNA 2005, 11, 371–382.
- Loll-Krippleber, R.; Brown, G.W. P-Body Proteins Regulate Transcriptional Rewiring to Promote DNA Replication Stress Resistance. Nat. Commun. 2017, 8, 558.
- Mugler, C.F.; Hondele, M.; Heinrich, S.; Sachdev, R.; Vallotton, P.; Koek, A.Y.; Chan, L.Y.; Weis, K. ATPase Activity of the DEAD-Box Protein Dhh1 Controls Processing Body Formation. Elife 2016, 5, e18746.
- Hondele, M.; Sachdev, R.; Heinrich, S.; Wang, J.; Vallotton, P.; Fontoura, B.M.A.; Weis, K. DEAD-Box ATPases Are Global Regulators of Phase-Separated Organelles. Nature 2019, 573, 144–148.
- Snead, W.T.; Gladfelter, A.S. The Control Centers of Biomolecular Phase Separation: How Membrane Surfaces, PTMs, and Active Processes Regulate Condensation. Mol. Cell 2019, 76, 295–305.
- Feric, M.; Vaidya, N.; Harmon, T.S.; Mitrea, D.M.; Zhu, L.; Richardson, T.M.; Kriwacki, R.W.; Pappu, R.V.; Brangwynne, C.P. Coexisting Liquid Phases Underlie Nucleolar Subcompartments. Cell 2016, 165, 1686–1697.
- Pullman, M.E.; Penefsky, H.S.; Datta, A.; Racker, E. Partial Resolution of the Enzymes Catalyzing Oxidative Phosphorylation. I. Purification and Properties of Soluble Dinitrophenol-Stimulated Adenosine Triphosphatase. J. Biol. Chem. 1960, 235, 3322–3329.
- Ernster, L.; Schatz, G. Mitochondria: A Historical Review. J. Cell Biol. 1981, 91, 227s–255s.
- Abrahams, J.P.; Leslie, A.G.; Lutter, R.; Walker, J.E. Structure at 2.8 A Resolution of F1-ATPase from Bovine Heart Mitochondria. Nature 1994, 370, 621–628.
- Grüber, G.; Wieczorek, H.; Harvey, W.R.; Müller, V. Structure–function Relationships of A-, F- and V-ATPases. J. Exp. Biol. 2001, 204, 2597–2605.
- Mitchell, P. Chemiosmotic Coupling in Oxidative and Photosynthetic Phosphorylation. Biol. Rev. Camb. Philos. Soc. 1966, 41, 445–502.
- Boyer, P.D. The ATP Synthase--a Splendid Molecular Machine. Annu. Rev. Biochem. 1997, 66, 717–749.
- Jonckheere, A.I.; Smeitink, J.A.M.; Rodenburg, R.J.T. Mitochondrial ATP Synthase: Architecture, Function and Pathology. J. Inherit. Metab. Dis. 2012, 35, 211–225.
- Strauss, M.; Hofhaus, G.; Schröder, R.R.; Kühlbrandt, W. Dimer Ribbons of ATP Synthase Shape the Inner Mitochondrial Membrane. EMBO J. 2008, 27, 1154–1160.
- Esparza-Perusquía, M.; Olvera-Sánchez, S.; Pardo, J.P.; Mendoza-Hernández, G.; Martínez, F.; Flores-Herrera, O. Structural and Kinetics Characterization of the F1F0-ATP Synthase Dimer. New Repercussion of Monomer-Monomer Contact. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 975–981.
- Spikes, T.E.; Montgomery, M.G.; Walker, J.E. Interface Mobility between Monomers in Dimeric Bovine ATP Synthase Participates in the Ultrastructure of Inner Mitochondrial Membranes. Proc. Natl. Acad. Sci. USA 2021, 118, e2021012118.
- Paumard, P.; Vaillier, J.; Coulary, B.; Schaeffer, J.; Soubannier, V.; Mueller, D.M.; Brèthes, D.; di Rago, J.-P.; Velours, J. The ATP Synthase Is Involved in Generating Mitochondrial Cristae Morphology. EMBO J. 2002, 21, 221–230.
- Davies, K.M.; Anselmi, C.; Wittig, I.; Faraldo-Gómez, J.D.; Kühlbrandt, W. Structure of the Yeast F1Fo-ATP Synthase Dimer and Its Role in Shaping the Mitochondrial Cristae. Proc. Natl. Acad. Sci. USA 2012, 109, 13602–13607.
- Habersetzer, J.; Larrieu, I.; Priault, M.; Salin, B.; Rossignol, R.; Brèthes, D.; Paumard, P. Human F1F0 ATP Synthase, Mitochondrial Ultrastructure and OXPHOS Impairment: A (super-)complex Matter? PLoS ONE 2013, 8, e75429.
- Mannella, C.A. The Relevance of Mitochondrial Membrane Topology to Mitochondrial Function. Biochim. Biophys. Acta 2006, 1762, 140–147.
- Blum, T.B.; Hahn, A.; Meier, T.; Davies, K.M.; Kühlbrandt, W. Dimers of Mitochondrial ATP Synthase Induce Membrane Curvature and Self-Assemble into Rows. Proc. Natl. Acad. Sci. USA 2019, 116, 4250–4255.
- Allen-Worthington, K.; Xie, J.; Brown, J.L.; Edmunson, A.M.; Dowling, A.; Navratil, A.M.; Scavelli, K.; Yoon, H.; Kim, D.-G.; Bynoe, M.S.; et al. The F0F1 ATP Synthase Complex Localizes to Membrane Rafts in Gonadotrope Cells. Mol. Endocrinol. 2016, 30, 996–1011.
- Kim, B.-W.; Lee, J.-W.; Choo, H.-J.; Lee, C.S.; Jung, S.-Y.; Yi, J.-S.; Ham, Y.-M.; Lee, J.-H.; Hong, J.; Kang, M.-J.; et al. Mitochondrial Oxidative Phosphorylation System Is Recruited to Detergent-Resistant Lipid Rafts during Myogenesis. Proteomics 2010, 10, 2498–2515.
- Hayes, M.H.; Peuchen, E.H.; Dovichi, N.J.; Weeks, D.L. Dual Roles for ATP in the Regulation of Phase Separated Protein Aggregates in Xenopus Oocyte Nucleoli. Elife 2018, 7, e35224.
- Sridharan, S.; Kurzawa, N.; Werner, T.; Günthner, I.; Helm, D.; Huber, W.; Bantscheff, M.; Savitski, M.M. Proteome-Wide Solubility and Thermal Stability Profiling Reveals Distinct Regulatory Roles for ATP. Nat. Commun. 2019, 10, 1155.
- Kuzmin, P.I.; Akimov, S.A.; Chizmadzhev, Y.A.; Zimmerberg, J.; Cohen, F.S. Line Tension and Interaction Energies of Membrane Rafts Calculated from Lipid Splay and Tilt. Biophys. J. 2005, 88, 1120–1133.
- Blackwell, D.J.; Zak, T.J.; Robia, S.L. Cardiac Calcium ATPase Dimerization Measured by Cross-Linking and Fluorescence Energy Transfer. Biophys. J. 2016, 111, 1192–1202.
- Houtkooper, R.H.; Vaz, F.M. Cardiolipin, the Heart of Mitochondrial Metabolism. Cell. Mol. Life Sci. 2008, 65, 2493–2506.
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438.
- Vähäheikkilä, M.; Peltomaa, T.; Róg, T.; Vazdar, M.; Pöyry, S.; Vattulainen, I. How Cardiolipin Peroxidation Alters the Properties of the Inner Mitochondrial Membrane? Chem. Phys. Lipids 2018, 214, 15–23.
- Sankhagowit, S.; Lee, E.Y.; Wong, G.C.L.; Malmstadt, N. Oxidation of Membrane Curvature-Regulating Phosphatidylethanolamine Lipid Results in Formation of Bilayer and Cubic Structures. Langmuir 2016, 32, 2450–2457.
- Acehan, D.; Malhotra, A.; Xu, Y.; Ren, M.; Stokes, D.L.; Schlame, M. Cardiolipin Affects the Supramolecular Organization of ATP Synthase in Mitochondria. Biophys. J. 2011, 100, 2184–2192.
- Reiter, R.J.; Rosales-Corral, S.; Tan, D.X.; Jou, M.J.; Galano, A.; Xu, B. Melatonin as a Mitochondria-Targeted Antioxidant: One of Evolution’s Best Ideas. Cell. Mol. Life Sci. 2017, 74, 3863–3881.
- Coon, S.L.; Klein, D.C. Evolution of Arylalkylamine N-Acetyltransferase: Emergence and Divergence. Mol. Cell. Endocrinol. 2006, 252, 2–10.
- Klein, D.C. Arylalkylamine N-Acetyltransferase: The Timezyme. J. Biol. Chem. 2007, 282, 4233–4237.
- Ganguly, S.; Weller, J.L.; Ho, A.; Chemineau, P.; Malpaux, B.; Klein, D.C. Melatonin Synthesis: 14-3-3-Dependent Activation and Inhibition of Arylalkylamine N-Acetyltransferase Mediated by Phosphoserine-205. Proc. Natl. Acad. Sci. USA 2005, 102, 1222–1227.
- Martín, M.; Macías, M.; Escames, G.; León, J.; Acuña-Castroviejo, D. Melatonin but Not Vitamins C and E Maintains Glutathione Homeostasis in T-Butyl Hydroperoxide-Induced Mitochondrial Oxidative Stress. FASEB J. 2000, 14, 1677–1679.
- Tan, D.-X.; Manchester, L.C.; Qin, L.; Reiter, R.J. Melatonin: A Mitochondrial Targeting Molecule Involving Mitochondrial Protection and Dynamics. Int. J. Mol. Sci. 2016, 17, 2124.
- Suofu, Y.; Li, W.; Jean-Alphonse, F.G.; Jia, J.; Khattar, N.K.; Li, J.; Baranov, S.V.; Leronni, D.; Mihalik, A.C.; He, Y.; et al. Dual Role of Mitochondria in Producing Melatonin and Driving GPCR Signaling to Block Cytochrome c Release. Proc. Natl. Acad. Sci. USA 2017, 114, E7997–E8006.
- Manchester, L.C.; Coto-Montes, A.; Boga, J.A.; Andersen, L.P.H.; Zhou, Z.; Galano, A.; Vriend, J.; Tan, D.-X.; Reiter, R.J. Melatonin: An Ancient Molecule That Makes Oxygen Metabolically Tolerable. J. Pineal. Res. 2015, 59, 403–419.
- Byeon, Y.; Lee, K.; Park, Y.-I.; Park, S.; Back, K. Molecular Cloning and Functional Analysis of Serotonin N-Acetyltransferase from the Cyanobacterium Synechocystis Sp. PCC 6803. J. Pineal. Res. 2013, 55, 371–376.
- Tan, D.-X.; Manchester, L.C.; Liu, X.; Rosales-Corral, S.A.; Acuna-Castroviejo, D.; Reiter, R.J. Mitochondria and Chloroplasts as the Original Sites of Melatonin Synthesis: A Hypothesis Related to Melatonin’s Primary Function and Evolution in Eukaryotes. J. Pineal. Res. 2013, 54, 127–138.
- Abhishek, A.; Bavishi, A.; Bavishi, A.; Choudhary, M. Bacterial Genome Chimaerism and the Origin of Mitochondria. Can. J. Microbiol. 2011, 57, 49–61.
- Raven, J.A.; Allen, J.F. Genomics and Chloroplast Evolution: What Did Cyanobacteria Do for Plants? Genome Biol. 2003, 4, 209.
- Tan, D.-X.; Manchester, L.C.; Terron, M.P.; Flores, L.J.; Reiter, R.J. One Molecule, Many Derivatives: A Never-Ending Interaction of Melatonin with Reactive Oxygen and Nitrogen Species? J. Pineal. Res. 2007, 42, 28–42.
- Liu, L.-N. Distribution and Dynamics of Electron Transport Complexes in Cyanobacterial Thylakoid Membranes. Biochim. Biophys. Acta 2016, 1857, 256–265.
- Gomes, E.; Shorter, J. The Molecular Language of Membraneless Organelles. J. Biol. Chem. 2019, 294, 7115–7127.
- Azaldegui, C.A.; Vecchiarelli, A.G.; Biteen, J.S. The Emergence of Phase Separation as an Organizing Principle in Bacteria. Biophys. J. 2021, 120, 1123–1138.
- Guilhas, B.; Walter, J.-C.; Rech, J.; David, G.; Walliser, N.O.; Palmeri, J.; Mathieu-Demaziere, C.; Parmeggiani, A.; Bouet, J.-Y.; Le Gall, A.; et al. ATP-Driven Separation of Liquid Phase Condensates in Bacteria. Mol. Cell 2020, 79, 293–303.e4.
- Muthunayake, N.S.; Tomares, D.T.; Childers, W.S.; Schrader, J.M. Phase-Separated Bacterial Ribonucleoprotein Bodies Organize mRNA Decay. Wiley Interdiscip. Rev. RNA 2020, 11, e1599.
- Oliver, T.; Sánchez-Baracaldo, P.; Larkum, A.W.; Rutherford, A.W.; Cardona, T. Time-Resolved Comparative Molecular Evolution of Oxygenic Photosynthesis. Biochim. Biophys. Acta Bioenerg. 2021, 1862, 148400.
- Pattanayak, G.K.; Liao, Y.; Wallace, E.W.J.; Budnik, B.; Drummond, D.A.; Rust, M.J. Daily Cycles of Reversible Protein Condensation in Cyanobacteria. Cell Rep. 2020, 32, 108032.
- Bar Eyal, L.; Ranjbar Choubeh, R.; Cohen, E.; Eisenberg, I.; Tamburu, C.; Dorogi, M.; Ünnep, R.; Appavou, M.-S.; Nevo, R.; Raviv, U.; et al. Changes in Aggregation States of Light-Harvesting Complexes as a Mechanism for Modulating Energy Transfer in Desert Crust Cyanobacteria. Proc. Natl. Acad. Sci. USA 2017, 114, 9481–9486.
- Wang, H.; Yan, X.; Aigner, H.; Bracher, A.; Nguyen, N.D.; Hee, W.Y.; Long, B.M.; Price, G.D.; Hartl, F.U.; Hayer-Hartl, M. Rubisco Condensate Formation by CcmM in β-Carboxysome Biogenesis. Nature 2019, 566, 131–135.
- McKinney, D.W.; Buchanan, B.B.; Wolosiuk, R.A. Activation of Chloroplast ATPase by Reduced Thioredoxin. Phytochemistry 1978, 17, 794–795.
- Curtis, S.E. Structure, Organization and Expression of Cyanobacterial ATP Synthase Genes. Photosynth. Res. 1988, 18, 223–244.
- Pogoryelov, D.; Reichen, C.; Klyszejko, A.L.; Brunisholz, R.; Muller, D.J.; Dimroth, P.; Meier, T. The Oligomeric State of c Rings from Cyanobacterial F-ATP Synthases Varies from 13 to 15. J. Bacteriol. 2007, 189, 5895–5902.
- Walraven, H.S.; Bakels, R.H.A. Function, Structure and Regulation of Cyanobacterial and Chloroplast ATP Synthase. Physiol. Plant 1996, 96, 526–532.
- Buchert, F.E. Chapter Three—Chloroplast ATP Synthase from Green Microalgae. In Advances in Botanical Research; Hisabori, T., Ed.; Academic Press: Cambridge, MA, USA, 2020; Volume 96, pp. 75–118.
- Carman, G.M. An Unusual Phosphatidylethanolamine-Utilizing Cardiolipin Synthase Is Discovered in Bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, 16402–16403.
- Kobayashi, K.; Endo, K.; Wada, H. Specific Distribution of Phosphatidylglycerol to Photosystem Complexes in the Thylakoid Membrane. Front. Plant Sci. 2017, 8, 1991.
- Basu Ball, W.; Neff, J.K.; Gohil, V.M. The Role of Nonbilayer Phospholipids in Mitochondrial Structure and Function. FEBS Lett. 2018, 592, 1273–1290.
- Shadyro, O.I.; Yurkova, I.L.; Kisel, M.A. Radiation-Induced Peroxidation and Fragmentation of Lipids in a Model Membrane. Int. J. Radiat. Biol. 2002, 78, 211–217.
- Althoff, T.; Mills, D.J.; Popot, J.-L.; Kühlbrandt, W. Arrangement of Electron Transport Chain Components in Bovine Mitochondrial Supercomplex I1III2IV1. EMBO J. 2011, 30, 4652–4664.
- Ostrander, D.B.; Zhang, M.; Mileykovskaya, E.; Rho, M.; Dowhan, W. Lack of Mitochondrial Anionic Phospholipids Causes an Inhibition of Translation of Protein Components of the Electron Transport Chain. A Yeast Genetic Model System for the Study of Anionic Phospholipid Function in Mitochondria. J. Biol. Chem. 2001, 276, 25262–25272.
- Yoshioka-Nishimura, M. Close Relationships Between the PSII Repair Cycle and Thylakoid Membrane Dynamics. Plant Cell Physiol. 2016, 57, 1115–1122.
- Megiatto, J.D.; Antoniuk-Pablant, A.; Sherman, B.D.; Kodis, G.; Gervaldo, M.; Moore, T.A.; Moore, A.L.; Gust, D. Mimicking the Electron Transfer Chain in Photosystem II with a Molecular Triad Thermodynamically Capable of Water Oxidation. Proc. Natl. Acad. Sci. USA 2012, 109, 15578–15583.
- Reiter, R.J. Melatonin: Lowering the High Price of Free Radicals. News Physiol. Sci. 2000, 15, 246–250.
- Reiter, R.J.; Tan, D.-X.; Terron, M.P.; Flores, L.J.; Czarnocki, Z. Melatonin and Its Metabolites: New Findings Regarding Their Production and Their Radical Scavenging Actions. Acta Biochim. Pol. 2007, 54, 1–9.
- Tan, D.X.; Manchester, L.C.; Reiter, R.J.; Plummer, B.F. Cyclic 3-Hydroxymelatonin: A Melatonin Metabolite Generated as a Result of Hydroxyl Radical Scavenging. Biol. Signals Recept. 1999, 8, 70–74.
- De Almeida, E.A.; Martinez, G.R.; Klitzke, C.F.; de Medeiros, M.H.G.; Di Mascio, P. Oxidation of Melatonin by Singlet Molecular Oxygen (O2(1deltag)) Produces N1-Acetyl-N2-Formyl-5-Methoxykynurenine. J. Pineal. Res. 2003, 35, 131–137.
- Matuszak, Z.; Bilska, M.A.; Reszka, K.J.; Chignell, C.F.; Bilski, P. Interaction of Singlet Molecular Oxygen with Melatonin and Related Indoles. Photochem. Photobiol. 2003, 78, 449–455.
- Tan, D.X.; Manchester, L.C.; Reiter, R.J.; Plummer, B.F.; Limson, J.; Weintraub, S.T.; Qi, W. Melatonin Directly Scavenges Hydrogen Peroxide: A Potentially New Metabolic Pathway of Melatonin Biotransformation. Free Radic. Biol. Med. 2000, 29, 1177–1185.
- Noda, Y.; Mori, A.; Liburdy, R.; Packer, L. Melatonin and Its Precursors Scavenge Nitric Oxide. J. Pineal. Res. 1999, 27, 159–163.
- Aydogan, S.; Yerer, M.B.; Goktas, A. Melatonin and Nitric Oxide. J. Endocrinol. Investig. 2006, 29, 281–287.
- Hardeland, R. Melatonin, Its Metabolites and Their Interference with Reactive Nitrogen Compounds. Molecules 2021, 26, 4105.
- Gilad, E.; Cuzzocrea, S.; Zingarelli, B.; Salzman, A.L.; Szabó, C. Melatonin Is a Scavenger of Peroxynitrite. Life Sci. 1997, 60, PL169–PL174.
- Galano, A.; Reiter, R.J. Melatonin and Its Metabolites vs. Oxidative Stress: From Individual Actions to Collective Protection. J. Pineal Res. 2018, 65, e12514.
- Purushothaman, A.; Sheeja, A.A.; Janardanan, D. Hydroxyl Radical Scavenging Activity of Melatonin and Its Related Indolamines. Free Radic. Res. 2020, 54, 373–383.
- Galano, A. On the Direct Scavenging Activity of Melatonin towards Hydroxyl and a Series of Peroxyl Radicals. Phys. Chem. Chem. Phys. 2011, 13, 7178–7188.
- Galano, A.; Tan, D.X.; Reiter, R.J. Cyclic 3-Hydroxymelatonin, a Key Metabolite Enhancing the Peroxyl Radical Scavenging Activity of Melatonin. RSC Adv. 2014, 4, 5220.
- Galano, A.; Medina, M.E.; Tan, D.X.; Reiter, R.J. Melatonin and Its Metabolites as Copper Chelating Agents and Their Role in Inhibiting Oxidative Stress: A Physicochemical Analysis. J. Pineal. Res. 2015, 58, 107–116.
- Lúcio, M.; Nunes, C.; Gaspar, D.; Ferreira, H.; Lima, J.L.F.C.; Reis, S. Antioxidant Activity of Vitamin E and Trolox: Understanding of the Factors That Govern Lipid Peroxidation Studies In Vitro. Food Biophys. 2009, 4, 312–320.
- Yu, H.; Dickson, E.J.; Jung, S.-R.; Koh, D.-S.; Hille, B. High Membrane Permeability for Melatonin. J. Gen. Physiol. 2016, 147, 63–76.
- Watson, H. Biological Membranes. Essays Biochem. 2015, 59, 43–69.
- Römsing, S. Development and Validation of Bioanalytical Methods: Application to Melatonin and Selected Anti-Infective Drugs; Acta Universitatis Upsaliensis: Uppsala, Sweden, 2010.
- Zhang, J.; Yan, X.; Tian, Y.; Li, W.; Wang, H.; Li, Q.; Li, Y.; Li, Z.; Wu, T. Synthesis of a New Water-Soluble Melatonin Derivative with Low Toxicity and a Strong Effect on Sleep Aid. ACS Omega. 2020, 5, 6494–6499.
- Shida, C.S.; Castrucci, A.M.; Lamy-Freund, M.T. High Melatonin Solubility in Aqueous Medium. J. Pineal. Res. 1994, 16, 198–201.
- Bongiorno, D.; Ceraulo, L.; Ferrugia, M.; Filizzola, F.; Giordano, C.; Ruggirello, A.; Liveri, V.T. H-NMR and FT-IR Study of the State of Melatonin Confined in Membrane Models: Location and Interactions of Melatonin in Water Free Lecithin and AOT Reversed Micelles. Arkivoc 2004, 2004, 251–262.
- Ceraulo, L.; Ferrugia, M.; Tesoriere, L.; Segreto, S.; Livrea, M.A.; Turco Liveri, V. Interactions of Melatonin with Membrane Models: Portioning of Melatonin in AOT and Lecithin Reversed Micelles. J. Pineal Res. 1999, 26, 108–112.
- Aikens, J.; Dix, T.A. Perhydroxyl Radical (HOO.) Initiated Lipid Peroxidation. The Role of Fatty Acid Hydroperoxides. J. Biol. Chem. 1991, 266, 15091–15098.
- Bielski, B.H.; Arudi, R.L.; Sutherland, M.W. A Study of the Reactivity of HO2/O2- with Unsaturated Fatty Acids. J. Biol. Chem. 1983, 258, 4759–4761.
- Repetto, M.; Semprine, J.; Boveris, A. Lipid Peroxidation: Chemical Mechanism, Biological Implications and Analytical Determination. In Lipid Peroxidation; Catala, A., Ed.; IntechOpen: London, UK, 2012.
- Ademowo, O.S.; Dias, H.K.I.; Burton, D.G.A.; Griffiths, H.R. Lipid (per) Oxidation in Mitochondria: An Emerging Target in the Ageing Process? Biogerontology 2017, 18, 859–879.
- Niki, E. Lipid Peroxidation: Physiological Levels and Dual Biological Effects. Free Radic. Biol. Med. 2009, 47, 469–484.
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and Biochemistry of 4-Hydroxynonenal, Malonaldehyde and Related Aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128.
- Kanner, J.; German, J.B.; Kinsella, J.E. Initiation of Lipid Peroxidation in Biological Systems. Crit. Rev. Food Sci. Nutr. 1987, 25, 317–364.
- Southorn, P.A.; Powis, G. Free Radicals in Medicine. I. Chemical Nature and Biologic Reactions. Mayo Clin. Proc. 1988, 63, 381–389.
- Yin, H.; Xu, L.; Porter, N.A. Free Radical Lipid Peroxidation: Mechanisms and Analysis. Chem. Rev. 2011, 111, 5944–5972.
- Wientjes, F.B.; Segal, A.W. NADPH Oxidase and the Respiratory Burst. Semin. Cell Biol. 1995, 6, 357–365.
- Wong, H.-S.; Dighe, P.A.; Mezera, V.; Monternier, P.-A.; Brand, M.D. Production of Superoxide and Hydrogen Peroxide from Specific Mitochondrial Sites under Different Bioenergetic Conditions. J. Biol. Chem. 2017, 292, 16804–16809.
- Bielski, B.H.J.; Cabelli, D.E.; Arudi, R.L.; Ross, A.B. Reactivity of HO2/O−2 Radicals in Aqueous Solution. J. Phys. Chem. Ref. Data 1985, 14, 1041–1100.
- Wardman, P. Reduction Potentials of One-Electron Couples Involving Free Radicals in Aqueous Solution. J. Phys. Chem. Ref. Data 1989, 18, 1637–1755.
- Hayyan, M.; Hashim, M.A.; AlNashef, I.M. Superoxide Ion: Generation and Chemical Implications. Chem. Rev. 2016, 116, 3029–3085.
- Collin, F. Chemical Basis of Reactive Oxygen Species Reactivity and Involvement in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 2407.
- Gebicki, J.M.; Bielski, B.H.J. Comparison of the Capacities of the Perhydroxyl and the Superoxide Radicals to Initiate Chain Oxidation of Linoleic Acid. J. Am. Chem. Soc. 1981, 103, 7020–7022.
- De Grey, A.D.N.J. HO2*: The Forgotten Radical. DNA Cell Biol. 2002, 21, 251–257.
- Halliwell, B.; Gutteridge, J.M. Oxygen Toxicity, Oxygen Radicals, Transition Metals and Disease. Biochem. J. 1984, 219, 1–14.
- Cordeiro, R.M. Reactive Oxygen Species at Phospholipid Bilayers: Distribution, Mobility and Permeation. Biochim. Biophys. Acta 2014, 1838, 438–444.
- Yusupov, M.; Wende, K.; Kupsch, S.; Neyts, E.C.; Reuter, S.; Bogaerts, A. Effect of Head Group and Lipid Tail Oxidation in the Cell Membrane Revealed through Integrated Simulations and Experiments. Sci. Rep. 2017, 7, 5761.
- Sathappa, M.; Alder, N.N. The Ionization Properties of Cardiolipin and Its Variants in Model Bilayers. Biochim. Biophys. Acta 2016, 1858, 1362–1372.
- Haines, T.H.; Dencher, N.A. Cardiolipin: A Proton Trap for Oxidative Phosphorylation. FEBS Lett. 2002, 528, 35–39.
- Horvath, S.E.; Daum, G. Lipids of Mitochondria. Prog. Lipid Res. 2013, 52, 590–614.
- Van den Brink-van der Laan, E.; Killian, J.A.; de Kruijff, B. Nonbilayer Lipids Affect Peripheral and Integral Membrane Proteins via Changes in the Lateral Pressure Profile. Biochim. Biophys. Acta 2004, 1666, 275–288.
- Khalifat, N.; Puff, N.; Bonneau, S.; Fournier, J.-B.; Angelova, M.I. Membrane Deformation under Local pH Gradient: Mimicking Mitochondrial Cristae Dynamics. Biophys. J. 2008, 95, 4924–4933.
- Parui, P.P.; Sarakar, Y.; Majumder, R.; Das, S.; Yang, H.; Yasuhara, K.; Hirota, S. Determination of Proton Concentration at Cardiolipin-Containing Membrane Interfaces and Its Relation with the Peroxidase Activity of Cytochrome c. Chem. Sci. 2019, 10, 9140–9151.
- Porcelli, A.M.; Ghelli, A.; Zanna, C.; Pinton, P.; Rizzuto, R.; Rugolo, M. pH Difference across the Outer Mitochondrial Membrane Measured with a Green Fluorescent Protein Mutant. Biochem. Biophys. Res. Commun. 2005, 326, 799–804.
- Afzal, N.; Lederer, W.J.; Jafri, M.S.; Mannella, C.A. Effect of Crista Morphology on Mitochondrial ATP Output: A Computational Study. Curr. Res. Physiol. 2021, 4, 163–176.
- Chicco, A.J.; Sparagna, G.C. Role of Cardiolipin Alterations in Mitochondrial Dysfunction and Disease. Am. J. Physiol. Cell Physiol. 2007, 292, C33–C44.
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Oxidative Stress, Cardiolipin and Mitochondrial Dysfunction in Nonalcoholic Fatty Liver Disease. World J. Gastroenterol. 2014, 20, 14205–14218.
- Kim, J.; Minkler, P.E.; Salomon, R.G.; Anderson, V.E.; Hoppel, C.L. Cardiolipin: Characterization of Distinct Oxidized Molecular Species. J. Lipid Res. 2011, 52, 125–135.
- Paradies, G.; Ruggiero, F.M.; Petrosillo, G.; Quagliariello, E. Age-Dependent Decline in the Cytochrome c Oxidase Activity in Rat Heart Mitochondria: Role of Cardiolipin. FEBS Lett. 1997, 406, 136–138.
- Paradies, G.; Ruggiero, F.M.; Petrosillo, G.; Quagliariello, E. Peroxidative Damage to Cardiac Mitochondria: Cytochrome Oxidase and Cardiolipin Alterations. FEBS Lett. 1998, 424, 155–158.
- Paradies, G.; Petrosillo, G.; Pistolese, M.; Ruggiero, F.M. Reactive Oxygen Species Generated by the Mitochondrial Respiratory Chain Affect the Complex III Activity via Cardiolipin Peroxidation in Beef-Heart Submitochondrial Particles. Mitochondrion 2001, 1, 151–159.
- Petrosillo, G.; Ruggiero, F.M.; Pistolese, M.; Paradies, G. Reactive Oxygen Species Generated from the Mitochondrial Electron Transport Chain Induce Cytochrome c Dissociation from Beef-Heart Submitochondrial Particles via Cardiolipin Peroxidation. Possible Role in the Apoptosis. FEBS Lett. 2001, 509, 435–438.
- Paradies, G.; Petrosillo, G.; Pistolese, M.; Ruggiero, F.M. Reactive Oxygen Species Affect Mitochondrial Electron Transport Complex I Activity through Oxidative Cardiolipin Damage. Gene 2002, 286, 135–141.
- Paradies, G.; Petrosillo, G.; Pistolese, M.; Di Venosa, N.; Federici, A.; Ruggiero, F.M. Decrease in Mitochondrial Complex I Activity in Ischemic/reperfused Rat Heart: Involvement of Reactive Oxygen Species and Cardiolipin. Circ. Res. 2004, 94, 53–59.
- Paradies, G.; Petrosillo, G.; Paradies, V.; Ruggiero, F.M. Role of Cardiolipin Peroxidation and Ca2+ in Mitochondrial Dysfunction and Disease. Cell Calcium. 2009, 45, 643–650.
- Arnarez, C.; Mazat, J.-P.; Elezgaray, J.; Marrink, S.-J.; Periole, X. Evidence for Cardiolipin Binding Sites on the Membrane-Exposed Surface of the Cytochrome bc1. J. Am. Chem. Soc. 2013, 135, 3112–3120.
- Panov, A. Perhydroxyl Radical (HO2•) as Inducer of the Isoprostane Lipid Peroxidation in Mitochondria. Mol. Biol. 2018, 52, 295–305.
- Miranda, É.G.A.; Araujo-Chaves, J.C.; Kawai, C.; Brito, A.M.M.; Dias, I.W.R.; Arantes, J.T.; Nantes-Cardoso, I.L. Cardiolipin Structure and Oxidation Are Affected by Ca2+ at the Interface of Lipid Bilayers. Front. Chem. 2019, 7, 930.
- Cipolla-Neto, J.; Amaral, F.G.; Afeche, S.C.; Tan, D.X.; Reiter, R.J. Melatonin, Energy Metabolism, and Obesity: A Review. J. Pineal. Res. 2014, 56, 371–381.
- Sustarsic, E.G.; Ma, T.; Lynes, M.D.; Larsen, M.; Karavaeva, I.; Havelund, J.F.; Nielsen, C.H.; Jedrychowski, M.P.; Moreno-Torres, M.; Lundh, M.; et al. Cardiolipin Synthesis in Brown and Beige Fat Mitochondria Is Essential for Systemic Energy Homeostasis. Cell Metab. 2018, 28, 159–174.e11.
- Von Bank, H.; Hurtado-Thiele, M.; Oshimura, N.; Simcox, J. Mitochondrial Lipid Signaling and Adaptive Thermogenesis. Metabolites 2021, 11, 124.
- Lee, Y.; Willers, C.; Kunji, E.R.S.; Crichton, P.G. Uncoupling Protein 1 Binds One Nucleotide per Monomer and Is Stabilized by Tightly Bound Cardiolipin. Proc. Natl. Acad. Sci. USA 2015, 112, 6973–6978.
- Fernández Vázquez, G.; Reiter, R.J.; Agil, A. Melatonin Increases Brown Adipose Tissue Mass and Function in Zücker Diabetic Fatty Rats: Implications for Obesity Control. J. Pineal. Res. 2018, 64, e12472.
- Elías-Wolff, F.; Lindén, M.; Lyubartsev, A.P.; Brandt, E.G. Curvature Sensing by Cardiolipin in Simulated Buckled Membranes. Soft Matter 2019, 15, 792–802.
- Ikon, N.; Ryan, R.O. Cardiolipin and Mitochondrial Cristae Organization. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1156–1163.
- Huang, Z.; Tyurina, Y.Y.; Jiang, J.; Tokarska-Schlattner, M.; Boissan, M.; Lacombe, M.-L.; Epand, R.; Schlattner, U.; Epand, R.M.; Kagan, V.E. Externalization of Cardiolipin as an “Eat-Me” Mitophageal Signal Is Facilitated by NDPK-D. Biophys. J. 2014, 106, 184a.
- Manganelli, V.; Capozzi, A.; Recalchi, S.; Riitano, G.; Mattei, V.; Longo, A.; Misasi, R.; Garofalo, T.; Sorice, M. The Role of Cardiolipin as a Scaffold Mitochondrial Phospholipid in Autophagosome Formation: In Vitro Evidence. Biomolecules 2021, 11, 222.
- Kagan, V.E.; Tyurin, V.A.; Jiang, J.; Tyurina, Y.Y.; Ritov, V.B.; Amoscato, A.A.; Osipov, A.N.; Belikova, N.A.; Kapralov, A.A.; Kini, V.; et al. Cytochrome c Acts as a Cardiolipin Oxygenase Required for Release of Proapoptotic Factors. Nat. Chem. Biol. 2005, 1, 223–232.
- Petrosillo, G.; Casanova, G.; Matera, M.; Ruggiero, F.M.; Paradies, G. Interaction of Peroxidized Cardiolipin with Rat-Heart Mitochondrial Membranes: Induction of Permeability Transition and Cytochrome c Release. FEBS Lett. 2006, 580, 6311–6316.
- Li, X.-X.; Tsoi, B.; Li, Y.-F.; Kurihara, H.; He, R.-R. Cardiolipin and Its Different Properties in Mitophagy and Apoptosis. J. Histochem. Cytochem. 2015, 63, 301–311.
- Dudek, J. Role of Cardiolipin in Mitochondrial Signaling Pathways. Front. Cell Dev. Biol. 2017, 5, 90.
- Huang, W.; Choi, W.; Hu, W.; Mi, N.; Guo, Q.; Ma, M.; Liu, M.; Tian, Y.; Lu, P.; Wang, F.-L.; et al. Crystal Structure and Biochemical Analyses Reveal Beclin 1 as a Novel Membrane Binding Protein. Cell Res. 2012, 22, 473–489.
- Gonzalvez, F.; Pariselli, F.; Dupaigne, P.; Budihardjo, I.; Lutter, M.; Antonsson, B.; Diolez, P.; Manon, S.; Martinou, J.-C.; Goubern, M.; et al. tBid Interaction with Cardiolipin Primarily Orchestrates Mitochondrial Dysfunctions and Subsequently Activates Bax and Bak. Cell Death Differ. 2005, 12, 614–626.
- Gonzalvez, F.; Schug, Z.T.; Houtkooper, R.H.; MacKenzie, E.D.; Brooks, D.G.; Wanders, R.J.A.; Petit, P.X.; Vaz, F.M.; Gottlieb, E. Cardiolipin Provides an Essential Activating Platform for Caspase-8 on Mitochondria. J. Cell Biol. 2008, 183, 681–696.
- Iyer, S.S.; He, Q.; Janczy, J.R.; Elliott, E.I.; Zhong, Z.; Olivier, A.K.; Sadler, J.J.; Knepper-Adrian, V.; Han, R.; Qiao, L.; et al. Mitochondrial Cardiolipin is Required for Nlrp3 Inflammasome Activation. Immunity 2013, 39, 311–323.
- De Zoete, M.R.; Palm, N.W.; Zhu, S.; Flavell, R.A. Inflammasomes. Cold Spring Harb. Perspect. Biol. 2014, 6, a016287.
- Sharif, H.; Wang, L.; Wang, W.L.; Magupalli, V.G.; Andreeva, L.; Qiao, Q.; Hauenstein, A.V.; Wu, Z.; Núñez, G.; Mao, Y.; et al. Structural Mechanism for NEK7-Licensed Activation of NLRP3 Inflammasome. Nature 2019, 570, 338–343.
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021.
- Seoane, P.I.; Lee, B.; Hoyle, C.; Yu, S.; Lopez-Castejon, G.; Lowe, M.; Brough, D. The NLRP3-Inflammasome as a Sensor of Organelle Dysfunction. J. Cell Biol. 2020, 219.
- Yeon, S.H.; Yang, G.; Lee, H.E.; Lee, J.Y. Oxidized Phosphatidylcholine Induces the Activation of NLRP3 Inflammasome in Macrophages. J. Leukoc. Biol. 2017, 101, 205–215.
- Elliott, E.I.; Miller, A.N.; Banoth, B.; Iyer, S.S.; Stotland, A.; Weiss, J.P.; Gottlieb, R.A.; Sutterwala, F.S.; Cassel, S.L. Cutting Edge: Mitochondrial Assembly of the NLRP3 Inflammasome Complex Is Initiated at Priming. J. Immunol. 2018, 200, 3047–3052.
- Raturi, A.; Simmen, T. Where the Endoplasmic Reticulum and the Mitochondrion Tie the Knot: The Mitochondria-Associated Membrane (MAM). Biochim. Biophys. Acta 2013, 1833, 213–224.
- Petrosillo, G.; Moro, N.; Ruggiero, F.M.; Paradies, G. Melatonin Inhibits Cardiolipin Peroxidation in Mitochondria and Prevents the Mitochondrial Permeability Transition and Cytochrome c Release. Free Radic. Biol. Med. 2009, 47, 969–974.
- Ono, K.; Mochizuki, H.; Ikeda, T.; Nihira, T.; Takasaki, J.-I.; Teplow, D.B.; Yamada, M. Effect of Melatonin on α-Synuclein Self-Assembly and Cytotoxicity. Neurobiol. Aging. 2012, 33, 2172–2185.
- Zampol, M.A.; Barros, M.H. Melatonin Improves Survival and Respiratory Activity of Yeast Cells Challenged by Alpha-Synuclein and Menadione. Yeast 2018, 35, 281–290.
- Samir, P.; Kanneganti, T.-D. DDX3X Sits at the Crossroads of Liquid-Liquid and Prionoid Phase Transitions Arbitrating Life and Death Cell Fate Decisions in Stressed Cells. DNA Cell Biol. 2020, 39, 1091–1095.
- Compan, V.; Martín-Sánchez, F.; Baroja-Mazo, A.; López-Castejón, G.; Gomez, A.I.; Verkhratsky, A.; Brough, D.; Pelegrín, P. Apoptosis-Associated Speck-like Protein Containing a CARD Forms Specks but Does Not Activate Caspase-1 in the Absence of NLRP3 during Macrophage Swelling. J. Immunol. 2015, 194, 1261–1273.
- Stehlik, C.; Lee, S.H.; Dorfleutner, A.; Stassinopoulos, A.; Sagara, J.; Reed, J.C. Apoptosis-Associated Speck-like Protein Containing a Caspase Recruitment Domain Is a Regulator of Procaspase-1 Activation. J. Immunol. 2003, 171, 6154–6163.
- Ozgur, S.; Buchwald, G.; Falk, S.; Chakrabarti, S.; Prabu, J.R.; Conti, E. The Conformational Plasticity of Eukaryotic RNA-Dependent ATPases. FEBS J. 2015, 282, 850–863.
- Owttrim, G.W. RNA Helicases: Diverse Roles in Prokaryotic Response to Abiotic Stress. RNA Biol. 2013, 10, 96–110.
- Aumiller, W.M.; Keating, C.D. Phosphorylation-Mediated RNA/peptide Complex Coacervation as a Model for Intracellular Liquid Organelles. Nat. Chem. 2016, 8, 129–137.
- Jin, M.; Fuller, G.G.; Han, T.; Yao, Y.; Alessi, A.F.; Freeberg, M.A.; Roach, N.P.; Moresco, J.J.; Karnovsky, A.; Baba, M.; et al. Glycolytic Enzymes Coalesce in G Bodies under Hypoxic Stress. Cell Rep. 2017, 20, 895–908.
- Kamagata, K.; Kanbayashi, S.; Honda, M.; Itoh, Y.; Takahashi, H.; Kameda, T.; Nagatsugi, F.; Takahashi, S. Liquid-like Droplet Formation by Tumor Suppressor p53 Induced by Multivalent Electrostatic Interactions between Two Disordered Domains. Sci. Rep. 2020, 10, 580.
- Hofweber, M.; Dormann, D. Friend or Foe-Post-Translational Modifications as Regulators of Phase Separation and RNP Granule Dynamics. J. Biol. Chem. 2019, 294, 7137–7150.
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Lo Muzio, L. The Crucial Role of Protein Phosphorylation in Cell Signaling and Its Use as Targeted Therapy (Review). Int. J. Mol. Med. 2017, 40, 271–280.
- Fox, D.; Man, S.M. DDX3X: Stressing the NLRP3 Inflammasome. Cell Res. 2019, 29, 969–970.
- Gustafson, E.A.; Wessel, G.M. DEAD-Box Helicases: Posttranslational Regulation and Function. Biochem. Biophys. Res. Commun. 2010, 395, 1–6.
- Soulat, D.; Bürckstümmer, T.; Westermayer, S.; Goncalves, A.; Bauch, A.; Stefanovic, A.; Hantschel, O.; Bennett, K.L.; Decker, T.; Superti-Furga, G. The DEAD-Box Helicase DDX3X Is a Critical Component of the TANK-Binding Kinase 1-Dependent Innate Immune Response. EMBO J. 2008, 27, 2135–2146.
- Ron, D. Translational Control in the Endoplasmic Reticulum Stress Response. J. Clin. Investig. 2002, 110, 1383–1388.
- Wek, R.C.; Jiang, H.-Y.; Anthony, T.G. Coping with Stress: eIF2 Kinases and Translational Control. Biochem. Soc. Trans. 2006, 34, 7–11.
- Donnelly, N.; Gorman, A.M.; Gupta, S.; Samali, A. The eIF2α Kinases: Their Structures and Functions. Cell. Mol. Life Sci. 2013, 70, 3493–3511.
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The Integrated Stress Response. EMBO Rep. 2016, 17, 1374–1395.
- Sidrauski, C.; McGeachy, A.M.; Ingolia, N.T.; Walter, P. The Small Molecule ISRIB Reverses the Effects of eIF2α Phosphorylation on Translation and Stress Granule Assembly. Elife 2015, 4, e05033.
- Wheeler, J.R.; Matheny, T.; Jain, S.; Abrisch, R.; Parker, R. Distinct Stages in Stress Granule Assembly and Disassembly. Elife 2016, 5, e18413.
- Anderson, P.; Kedersha, N. Visibly Stressed: The Role of eIF2, TIA-1, and Stress Granules in Protein Translation. Cell Stress Chaperones 2002, 7, 213–221.
- Miller, Y.I.; Navia-Pelaez, J.M.; Corr, M.; Yaksh, T.L. Lipid Rafts in Glial Cells: Role in Neuroinflammation and Pain Processing: Thematic Review Series: Biology of Lipid Rafts. J. Lipid Res. 2020, 61, 655–666.
- Li, Y.C.; Park, M.J.; Ye, S.-K.; Kim, C.-W.; Kim, Y.-N. Elevated Levels of Cholesterol-Rich Lipid Rafts in Cancer Cells Are Correlated with Apoptosis Sensitivity Induced by Cholesterol-Depleting Agents. Am. J. Pathol. 2006, 168, 1107–1118; quiz 1404–1405.
- Guan, Y.; Han, F. Key Mechanisms and Potential Targets of the NLRP3 Inflammasome in Neurodegenerative Diseases. Front. Integr. Neurosci. 2020, 14, 37.
- Arioz, B.I.; Tastan, B.; Tarakcioglu, E.; Tufekci, K.U.; Olcum, M.; Ersoy, N.; Bagriyanik, A.; Genc, K.; Genc, S. Melatonin Attenuates LPS-Induced Acute Depressive-Like Behaviors and Microglial NLRP3 Inflammasome Activation Through the SIRT1/Nrf2 Pathway. Front. Immunol. 2019, 10, 1511.
- Zhang, J.; Lu, X.; Liu, M.; Fan, H.; Zheng, H.; Zhang, S.; Rahman, N.; Wołczyński, S.; Kretowski, A.; Li, X. Melatonin Inhibits Inflammasome-Associated Activation of Endothelium and Macrophages Attenuating Pulmonary Arterial Hypertension. Cardiovasc. Res. 2020, 116, 2156–2169.
- Chen, F.; Jiang, G.; Liu, H.; Li, Z.; Pei, Y.; Wang, H.; Pan, H.; Cui, H.; Long, J.; Wang, J.; et al. Melatonin Alleviates Intervertebral Disc Degeneration by Disrupting the IL-1β/NF-κB-NLRP3 Inflammasome Positive Feedback Loop. Bone Res. 2020, 8, 10.
- Severcan, F.; Sahin, I.; Kazanci, N. Melatonin Strongly Interacts with Zwitterionic Model Membranes--Evidence from Fourier Transform Infrared Spectroscopy and Differential Scanning Calorimetry. Biochim. Biophys. Acta 2005, 1668, 215–222.
- Wong-Ekkabut, J.; Xu, Z.; Triampo, W.; Tang, I.-M.; Tieleman, D.P.; Monticelli, L. Effect of Lipid Peroxidation on the Properties of Lipid Bilayers: A Molecular Dynamics Study. Biophys. J. 2007, 93, 4225–4236.
- Kaplán, P.; Racay, P.; Lehotský, J.; Mézesová, V. Change in Fluidity of Brain Endoplasmic Reticulum Membranes by Oxygen Free Radicals: A Protective Effect of Stobadine, Alpha-Tocopherol Acetate, and Butylated Hydroxytoluene. Neurochem. Res. 1995, 20, 815–820.
- Yu, B.P.; Suescun, E.A.; Yang, S.Y. Effect of Age-Related Lipid Peroxidation on Membrane Fluidity and Phospholipase A2: Modulation by Dietary Restriction. Mech. Ageing. Dev. 1992, 65, 17–33.
- Dies, H.; Toppozini, L.; Rheinstädter, M.C. The Interaction between Amyloid-β Peptides and Anionic Lipid Membranes Containing Cholesterol and Melatonin. PLoS ONE 2014, 9, e99124.
- Dies, H.; Cheung, B.; Tang, J.; Rheinstädter, M.C. The Organization of Melatonin in Lipid Membranes. Biochim. Biophys. Acta 2015, 1848, 1032–1040.
- Rudzite, V.; Jurika, E.; Jirgensons, J. Changes in Membrane Fluidity Induced by Tryptophan and Its Metabolites. In Tryptophan, Serotonin, and Melatonin: Basic Aspects and Applications; Huether, G., Kochen, W., Simat, T.J., Steinhart, H., Eds.; Springer: Boston, MA, USA, 1999.
- Beyenbach, K.W.; Wieczorek, H. The V-Type H+ ATPase: Molecular Structure and Function, Physiological Roles and Regulation. J. Exp. Biol. 2006, 209 Pt 4, 577–589.
- Bae, T.-J.; Kim, M.-S.; Kim, J.-W.; Kim, B.-W.; Choo, H.-J.; Lee, J.-W.; Kim, K.-B.; Lee, C.S.; Kim, J.-H.; Chang, S.Y.; et al. Lipid Raft Proteome Reveals ATP Synthase Complex in the Cell Surface. Proteomics 2004, 4, 3536–3548.
- Kim, B.-W.; Choo, H.-J.; Lee, J.-W.; Kim, J.-H.; Ko, Y.-G. Extracellular ATP Is Generated by ATP Synthase Complex in Adipocyte Lipid Rafts. Exp. Mol. Med. 2004, 36, 476–485.
- Alexandre, H.; Mathieu, B.; Charpentier, C. Alteration in Membrane Fluidity and Lipid Composition, and Modulation of H+-ATPase Activity in Saccharomyces Cerevisiae Caused by Decanoic Acid. Available online: https://www.microbiologyresearch.org/docserver/fulltext/micro/142/3/mic-142-3-469.pdf?expires=1616871084&id=id&accname=guest&checksum=6688BACF19B736B01B5100ACDEA617F6 (accessed on 27 March 2021).
- Keeffe, E.B.; Blankenship, N.M.; Scharschmidt, B.F. Alteration of Rat Liver Plasma Membrane Fluidity and ATPase Activity by Chlorpromazine Hydrochloride and Its Metabolites. Gastroenterology 1980, 79, 222–231.
- Garcia, A.; Pochinda, S.; Elgaard-Jørgensen, P.N.; Khandelia, H.; Clarke, R.J. Evidence for ATP Interaction with Phosphatidylcholine Bilayers. Langmuir 2019, 35, 9944–9953.
- Chen, J.J.; Yu, B.P. Alterations in Mitochondrial Membrane Fluidity by Lipid Peroxidation Products. Free Radic. Biol. Med. 1994, 17, 411–418.
- Kholodenko, B.N.; Hoek, J.B.; Westerhoff, H.V. Why Cytoplasmic Signalling Proteins Should Be Recruited to Cell Membranes. Trends Cell Biol. 2000, 10, 173–178.
- Botterbusch, S.; Baumgart, T. Interactions between Phase-Separated Liquids and Membrane Surfaces. NATO Adv. Sci. Inst. Ser. E Appl. Sci. 2021, 11, 1288.
- Alimohamadi, H.; Rangamani, P. Modeling Membrane Curvature Generation due to Membrane−Protein Interactions. Biomolecules 2018, 8, 120.
- Prévost, C.; Zhao, H.; Manzi, J.; Lemichez, E.; Lappalainen, P.; Callan-Jones, A.; Bassereau, P. IRSp53 Senses Negative Membrane Curvature and Phase Separates along Membrane Tubules. Nat. Commun. 2015, 6, 8529.
- Gallop, J.L.; McMahon, H.T. BAR Domains and Membrane Curvature: Bringing Your Curves to the BAR. Biochem. Soc. Symp. 2005, 72, 223–231.
- Fabiani, C.; Antollini, S.S. Alzheimer’s Disease as a Membrane Disorder: Spatial Cross-Talk Among Beta-Amyloid Peptides, Nicotinic Acetylcholine Receptors and Lipid Rafts. Front. Cell Neurosci. 2019, 13, 309.
- Lu, S.; Deng, R.; Jiang, H.; Song, H.; Li, S.; Shen, Q.; Huang, W.; Nussinov, R.; Yu, J.; Zhang, J. The Mechanism of ATP-Dependent Allosteric Protection of Akt Kinase Phosphorylation. Structure 2015, 23, 1725–1734.
- Simons, K.; Ikonen, E. Functional Rafts in Cell Membranes. Nature 1997, 387, 569–572.
- Simons, K.; Toomre, D. Lipid Rafts and Signal Transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39.
- Bagnat, M.; Keränen, S.; Shevchenko, A.; Shevchenko, A.; Simons, K. Lipid Rafts Function in Biosynthetic Delivery of Proteins to the Cell Surface in Yeast. Proc. Natl. Acad. Sci. USA 2000, 97, 3254–3259.
- Ikonen, E. Roles of Lipid Rafts in Membrane Transport. Curr. Opin. Cell Biol. 2001, 13, 470–477.
- Jin, S.; Zhou, F.; Katirai, F.; Li, P.-L. Lipid Raft Redox Signaling: Molecular Mechanisms in Health and Disease. Antioxid. Redox Signal. 2011, 15, 1043–1083.
- Jiang, F.; Zhang, Y.; Dusting, G.J. NADPH Oxidase-Mediated Redox Signaling: Roles in Cellular Stress Response, Stress Tolerance, and Tissue Repair. Pharmacol. Rev. 2011, 63, 218–242.
- Garofalo, T.; Manganelli, V.; Grasso, M.; Mattei, V.; Ferri, A.; Misasi, R.; Sorice, M. Role of Mitochondrial Raft-like Microdomains in the Regulation of Cell Apoptosis. Apoptosis 2015, 20, 621–634.
- Samhan-Arias, A.K.; Garcia-Bereguiain, M.A.; Martin-Romero, F.J.; Gutierrez-Merino, C. Clustering of Plasma Membrane-Bound Cytochrome b5 Reductase within “Lipid Raft” Microdomains of the Neuronal Plasma Membrane. Mol. Cell. Neurosci. 2009, 40, 14–26.
- Kinnun, J.J.; Bolmatov, D.; Lavrentovich, M.O.; Katsaras, J. Lateral Heterogeneity and Domain Formation in Cellular Membranes. Chem. Phys. Lipids 2020, 232, 104976.
- Heerklotz, H. Triton Promotes Domain Formation in Lipid Raft Mixtures. Biophys. J. 2002, 83, 2693–2701.
- Bolmatov, D.; Soloviov, D.; Zhernenkov, M.; Zav’yalov, D.; Mamontov, E.; Suvorov, A.; Cai, Y.Q.; Katsaras, J. Molecular Picture of the Transient Nature of Lipid Rafts. Langmuir 2020, 36, 4887–4896.
- Aponte-Santamaría, C.; Brunken, J.; Gräter, F. Stress Propagation through Biological Lipid Bilayers in Silico. J. Am. Chem. Soc. 2017, 139, 13588–13591.
- Mei, N.; Robinson, M.; Davis, J.H.; Leonenko, Z. Melatonin Alters Fluid Phase Co-Existence in POPC/DPPC/cholesterol Membranes. Biophys. J. 2020, 119, 2391–2402.
- Choi, Y.; Attwood, S.J.; Hoopes, M.I.; Drolle, E.; Karttunen, M.; Leonenko, Z. Melatonin Directly Interacts with Cholesterol and Alleviates Cholesterol Effects in Dipalmitoylphosphatidylcholine Monolayers. Soft Matter 2014, 10, 206–213.
- Del Mar Martínez-Senac, M.; Villalaín, J.; Gómez-Fernández, J.C. Structure of the Alzheimer Beta-Amyloid Peptide (25-35) and Its Interaction with Negatively Charged Phospholipid Vesicles. Eur. J. Biochem. 1999, 265, 744–753.
- Sani, M.-A.; Gehman, J.D.; Separovic, F. Lipid Matrix Plays a Role in Abeta Fibril Kinetics and Morphology. FEBS Lett. 2011, 585, 749–754.
- Hane, F.; Drolle, E.; Gaikwad, R.; Faught, E.; Leonenko, Z. Amyloid-β Aggregation on Model Lipid Membranes: An Atomic Force Microscopy Study. J. Alzheimers. Dis. 2011, 26, 485–494.
- Ahyayauch, H.; Raab, M.; Busto, J.V.; Andraka, N.; Arrondo, J.-L.R.; Masserini, M.; Tvaroska, I.; Goñi, F.M. Binding of β-Amyloid (1-42) Peptide to Negatively Charged Phospholipid Membranes in the Liquid-Ordered State: Modeling and Experimental Studies. Biophys. J. 2012, 103, 453–463.
- Ding, H.; Schauerte, J.A.; Steel, D.G.; Gafni, A. β-Amyloid (1-40) Peptide Interactions with Supported Phospholipid Membranes: A Single-Molecule Study. Biophys. J. 2012, 103, 1500–1509.
- Magarkar, A.; Dhawan, V.; Kallinteri, P.; Viitala, T.; Elmowafy, M.; Róg, T.; Bunker, A. Cholesterol Level Affects Surface Charge of Lipid Membranes in Saline Solution. Sci. Rep. 2014, 4, 5005.
- Chen, Z.; Rand, R.P. The Influence of Cholesterol on Phospholipid Membrane Curvature and Bending Elasticity. Biophys. J. 1997, 73, 267–276.
- Drolle, E.; Gaikwad, R.M.; Leonenko, Z. Nanoscale Electrostatic Domains in Cholesterol-Laden Lipid Membranes Create a Target for Amyloid Binding. Biophys. J. 2012, 103, L27–L29.
- Finot, E.; Leonenko, Y.; Moores, B.; Eng, L.; Amrein, M.; Leonenko, Z. Effect of Cholesterol on Electrostatics in Lipid-Protein Films of a Pulmonary Surfactant. Langmuir 2010, 26, 1929–1935.
- Eckert, G.P.; Kirsch, C.; Leutz, S.; Wood, W.G.; Müller, W.E. Cholesterol Modulates Amyloid Beta-Peptide’s Membrane Interactions. Pharmacopsychiatry 2003, 36 (Suppl. 2), S136–S143.
- Gibson Wood, W.; Eckert, G.P.; Igbavboa, U.; Müller, W.E. Amyloid Beta-Protein Interactions with Membranes and Cholesterol: Causes or Casualties of Alzheimer’s Disease. Biochim. Biophys. Acta 2003, 1610, 281–290.
- Gostincar, C.; Turk, M.; Gunde-Cimerman, N. The Evolution of Fatty Acid Desaturases and Cytochrome b5 in Eukaryotes. J. Membr. Biol. 2010, 233, 63–72.
- Ito, A.; Hayashi, S.; Yoshida, T. Participation of a Cytochrome b5-like Hemoprotein of Outer Mitochondrial Membrane (OM Cytochrome B) in NADH-Semidehydroascorbic Acid Reductase Activity of Rat Liver. Biochem. Biophys. Res. Commun. 1981, 101, 591–598.
- Navarro, F.; Villalba, J.M.; Crane, F.L.; Mackellar, W.C.; Navas, P. A Phospholipid-Dependent NADH-Coenzyme Q Reductase from Liver Plasma Membrane. Biochem. Biophys. Res. Commun. 1995, 212, 138–143.
- Percy, M.J.; Lappin, T.R. Recessive Congenital Methaemoglobinaemia: Cytochrome b(5) Reductase Deficiency. Br. J. Haematol. 2008, 141, 298–308.
- Siendones, E.; SantaCruz-Calvo, S.; Martín-Montalvo, A.; Cascajo, M.V.; Ariza, J.; López-Lluch, G.; Villalba, J.M.; Acquaviva-Bourdain, C.; Roze, E.; Bernier, M.; et al. Membrane-Bound CYB5R3 Is a Common Effector of Nutritional and Oxidative Stress Response through FOXO3a and Nrf2. Antioxid. Redox Signal. 2014, 21, 1708–1725.
- Marques-da-Silva, D.; Samhan-Arias, A.K.; Tiago, T.; Gutierrez-Merino, C. L-Type Calcium Channels and Cytochrome b5 Reductase Are Components of Protein Complexes Tightly Associated with Lipid Rafts Microdomains of the Neuronal Plasma Membrane. J. Proteom. 2010, 73, 1502–1510.
- Nikiforova, A.B.; Saris, N.-E.L.; Kruglov, A.G. External Mitochondrial NADH-Dependent Reductase of Redox Cyclers: VDAC1 or Cyb5R3? Free Radic. Biol. Med. 2014, 74, 74–84.
- Bakalova, R.; Zhelev, Z.; Miller, T.; Aoki, I.; Higashi, T. New Potential Biomarker for Stratification of Patients for Pharmacological Vitamin C in Adjuvant Settings of Cancer Therapy. Redox Biol. 2020, 28, 101357.
- Mihara, K.; Sato, R. Molecular Cloning and Sequencing of cDNA for Yeast Porin, an Outer Mitochondrial Membrane Protein: A Search for Targeting Signal in the Primary Structure. EMBO J. 1985, 4, 769–774.
- Thinnes, F.P.; Götz, H.; Kayser, H.; Benz, R.; Schmidt, W.E.; Kratzin, H.D.; Hilschmann, N. Identification of human porins. I. Purification of a porin from human B-lymphocytes (Porin 31HL) and the topochemical proof of its expression on the plasmalemma of the progenitor cell. Biol. Chem. Hoppe Seyler 1989, 370, 1253–1264.
- Bàthori, G.; Parolini, I.; Tombola, F.; Szabò, I.; Messina, A.; Oliva, M.; De Pinto, V.; Lisanti, M.; Sargiacomo, M.; Zoratti, M. Porin Is Present in the Plasma Membrane Where It Is Concentrated in Caveolae and Caveolae-Related Domains. J. Biol. Chem. 1999, 274, 29607–29612.
- Herrera, J.L.; Diaz, M.; Hernández-Fernaud, J.R.; Salido, E.; Alonso, R.; Fernández, C.; Morales, A.; Marin, R. Voltage-Dependent Anion Channel as a Resident Protein of Lipid Rafts: Post-Transductional Regulation by Estrogens and Involvement in Neuronal Preservation against Alzheimer’s Disease. J. Neurochem. 2011, 116, 820–827.
- De Pinto, V.; Messina, A.; Lane, D.J.R.; Lawen, A. Voltage-Dependent Anion-Selective Channel (VDAC) in the Plasma Membrane. FEBS Lett. 2010, 584, 1793–1799.
- Anishkin, A.; Loukin, S.H.; Teng, J.; Kung, C. Feeling the Hidden Mechanical Forces in Lipid Bilayer Is an Original Sense. Proc. Natl. Acad. Sci. USA 2014, 111, 7898–7905.
- Anishkin, A.; Kung, C. Stiffened Lipid Platforms at Molecular Force Foci. Proc. Natl. Acad. Sci. USA 2013, 110, 4886–4892.
- Martinac, B.; Adler, J.; Kung, C. Mechanosensitive Ion Channels of E. Coli Activated by Amphipaths. Nature 1990, 348, 261–263.
- Markin, V.S.; Martinac, B. Mechanosensitive Ion Channels as Reporters of Bilayer Expansion. A Theoretical Model. Biophys. J. 1991, 60, 1120–1127.
- Dart, C. Lipid Microdomains and the Regulation of Ion Channel Function. J. Physiol. 2010, 588 Pt 17, 3169–3178.
- Samhan-Arias, A.K.; Marques-da-Silva, D.; Yanamala, N.; Gutierrez-Merino, C. Stimulation and Clustering of Cytochrome b5 Reductase in Caveolin-Rich Lipid Microdomains Is an Early Event in Oxidative Stress-Mediated Apoptosis of Cerebellar Granule Neurons. J. Proteom. 2012, 75, 2934–2949.
- Samhan-Arias, A.K.; Fortalezas, S.; Cordas, C.M.; Moura, I.; Moura, J.J.G.; Gutierrez-Merino, C. Cytochrome b5 Reductase Is the Component from Neuronal Synaptic Plasma Membrane Vesicles That Generates Superoxide Anion upon Stimulation by Cytochrome c. Redox Biol. 2018, 15, 109–114.
- Samhan-Arias, A.K.; Gutierrez-Merino, C. Purified NADH-Cytochrome b5 Reductase Is a Novel Superoxide Anion Source Inhibited by Apocynin: Sensitivity to Nitric Oxide and Peroxynitrite. Free Radic. Biol. Med. 2014, 73, 174–189.
- La Piana, G.; Marzulli, D.; Gorgoglione, V.; Lofrumento, N.E. Porin and Cytochrome Oxidase Containing Contact Sites Involved in the Oxidation of Cytosolic NADH. Arch. Biochem. Biophys. 2005, 436, 91–100.
- Martín-Romero, F.J.; Gutiérrez-Martín, Y.; Henao, F.; Gutiérrez-Merino, C. The NADH Oxidase Activity of the Plasma Membrane of Synaptosomes Is a Major Source of Superoxide Anion and Is Inhibited by Peroxynitrite. J. Neurochem. 2002, 82, 604–614.
- Zizi, M.; Forte, M.; Blachly-Dyson, E.; Colombini, M. NADH Regulates the Gating of VDAC, the Mitochondrial Outer Membrane Channel. J. Biol. Chem. 1994, 269, 1614–1616.
- Shoshan-Barmatz, V.; Shteinfer-Kuzmine, A.; Verma, A. VDAC1 at the Intersection of Cell Metabolism, Apoptosis, and Diseases. Biomolecules 2020, 10, 1485.
- Lemasters, J.J. Evolution of Voltage-Dependent Anion Channel Function: From Molecular Sieve to Governator to Actuator of Ferroptosis. Front. Oncol. 2017, 7, 303.
- Elinder, F.; Akanda, N.; Tofighi, R.; Shimizu, S.; Tsujimoto, Y.; Orrenius, S.; Ceccatelli, S. Opening of Plasma Membrane Voltage-Dependent Anion Channels (VDAC) Precedes Caspase Activation in Neuronal Apoptosis Induced by Toxic Stimuli. Cell Death Differ. 2005, 12, 1134–1140.
- Rostovtseva, T.; Colombini, M. VDAC Channels Mediate and Gate the Flow of ATP: Implications for the Regulation of Mitochondrial Function. Biophys. J. 1997, 72, 1954–1962.
- Rostovtseva, T.K.; Tan, W.; Colombini, M. On the Role of VDAC in Apoptosis: Fact and Fiction. J. Bioenerg. Biomembr. 2005, 37, 129–142.
- McCommis, K.S.; Baines, C.P. The Role of VDAC in Cell Death: Friend or Foe? Biochim. Biophys. Acta 2012, 1818, 1444–1450.
- Shoshan-Barmatz, V.; Maldonado, E.N.; Krelin, Y. VDAC1 at the Crossroads of Cell Metabolism, Apoptosis and Cell Stress. Cell Stress Chaperones 2017, 1, 11–36.
- Camara, A.K.S.; Zhou, Y.; Wen, P.-C.; Tajkhorshid, E.; Kwok, W.-M. Mitochondrial VDAC1: A Key Gatekeeper as Potential Therapeutic Target. Front. Physiol. 2017, 8, 460.
- Sorice, M.; Manganelli, V.; Matarrese, P.; Tinari, A.; Misasi, R.; Malorni, W.; Garofalo, T. Cardiolipin-Enriched Raft-like Microdomains Are Essential Activating Platforms for Apoptotic Signals on Mitochondria. FEBS Lett. 2009, 583, 2447–2450.
- Sorice, M.; Mattei, V.; Matarrese, P.; Garofalo, T.; Tinari, A.; Gambardella, L.; Ciarlo, L.; Manganelli, V.; Tasciotti, V.; Misasi, R.; et al. Dynamics of Mitochondrial Raft-like Microdomains in Cell Life and Death. Commun. Integr. Biol. 2012, 5, 217–219.
- Malorni, W.; Giammarioli, A.M.; Garofalo, T.; Sorice, M. Dynamics of Lipid Raft Components during Lymphocyte Apoptosis: The Paradigmatic Role of GD3. Apoptosis 2007, 12, 941–949.
- Brand, M.D. Mitochondrial Generation of Superoxide and Hydrogen Peroxide as the Source of Mitochondrial Redox Signaling. Free Radic. Biol. Med. 2016, 100, 14–31.
- Schneider, C. An Update on Products and Mechanisms of Lipid Peroxidation. Mol. Nutr. Food Res. 2009, 53, 315–321.
- Ting, H.-C.; Chen, L.-T.; Chen, J.-Y.; Huang, Y.-L.; Xin, R.-C.; Chan, J.-F.; Hsu, Y.-H.H. Double Bonds of Unsaturated Fatty Acids Differentially Regulate Mitochondrial Cardiolipin Remodeling. Lipids Health Dis. 2019, 18, 53.
- Musatov, A. Contribution of Peroxidized Cardiolipin to Inactivation of Bovine Heart Cytochrome c Oxidase. Free Radic. Biol. Med. 2006, 41, 238–246.
- Venegas, C.; García, J.A.; Escames, G.; Ortiz, F.; López, A.; Doerrier, C.; García-Corzo, L.; López, L.C.; Reiter, R.J.; Acuña-Castroviejo, D. Extrapineal Melatonin: Analysis of Its Subcellular Distribution and Daily Fluctuations. J. Pineal Res. 2012, 52, 217–227.
- Cesarini, E.; Cerioni, L.; Canonico, B.; Di Sario, G.; Guidarelli, A.; Lattanzi, D.; Savelli, D.; Guescini, M.; Nasoni, M.G.; Bigini, N.; et al. Melatonin Protects Hippocampal HT22 Cells from the Effects of Serum Deprivation Specifically Targeting Mitochondria. PLoS ONE 2018, 13, e0203001.
- Mookerjee, S.A.; Gerencser, A.A.; Nicholls, D.G.; Brand, M.D. Quantifying Intracellular Rates of Glycolytic and Oxidative ATP Production and Consumption Using Extracellular Flux Measurements. J. Biol. Chem. 2017, 292, 7189–7207.
- Martín, M.; Macías, M.; León, J.; Escames, G.; Khaldy, H.; Acuña-Castroviejo, D. Melatonin Increases the Activity of the Oxidative Phosphorylation Enzymes and the Production of ATP in Rat Brain and Liver Mitochondria. Int. J. Biochem. Cell Biol. 2002, 34, 348–357.
- Chen, X.; Hao, B.; Li, D.; Reiter, R.J.; Bai, Y.; Abay, B.; Chen, G.; Lin, S.; Zheng, T.; Ren, Y.; et al. Melatonin Inhibits Lung Cancer Development by Reversing the Warburg Effect via Stimulating the SIRT3/PDH Axis. J. Pineal. Res. 2021, e12755.
- Reiter, R.J.; Sharma, R.; Rosales-Corral, S. Anti-Warburg Effect of Melatonin: A Proposed Mechanism to Explain Its Inhibition of Multiple Diseases. Int. J. Mol. Sci. 2021, 22, 764.
- Reiter, R.J.; Sharma, R.; Ma, Q.; Rorsales-Corral, S.; de Almeida Chuffa, L.G. Melatonin Inhibits Warburg-Dependent Cancer by Redirecting Glucose Oxidation to the Mitochondria: A Mechanistic Hypothesis. Cell. Mol. Life Sci. 2020, 77, 2527–2542.
- Reiter, R.J.; Sharma, R.; Pires de Campos Zuccari, D.A.; de Almeida Chuffa, L.G.; Manucha, W.; Rodriguez, C. Melatonin Synthesis in and Uptake by Mitochondria: Implications for Diseased Cells with Dysfunctional Mitochondria. Future Med. Chem. 2021, 13, 335–339.
- Xia, Y.; Chen, S.; Zeng, S.; Zhao, Y.; Zhu, C.; Deng, B.; Zhu, G.; Yin, Y.; Wang, W.; Hardeland, R.; et al. Melatonin in Macrophage Biology: Current Understanding and Future Perspectives. J. Pineal. Res. 2019, 66, e12547.
- Reiter, R.J.; Sharma, R.; Ma, Q. Switching Diseased Cells from Cytosolic Aerobic Glycolysis to Mitochondrial Oxidative Phosphorylation: A Metabolic Rhythm Regulated by Melatonin? J. Pineal. Res. 2021, 70, e12677.
- Fuller, G.G.; Han, T.; Freeberg, M.A.; Moresco, J.J.; Ghanbari Niaki, A.; Roach, N.P.; Yates, J.R.; Myong, S.; Kim, J.K. RNA Promotes Phase Separation of Glycolysis Enzymes into Yeast G Bodies in Hypoxia. Elife 2020, 9, e48480.
- Sarkar, S.; Mondal, J. Mechanistic Insights on ATP’s Role as a Hydrotrope. J. Phys. Chem. B 2021, 125, 7717–7731.
- Maldonado, E.N.; Lemasters, J.J. ATP/ADP Ratio, the Missed Connection between Mitochondria and the Warburg Effect. Mitochondrion 2014, 19 Pt A, 78–84.
- Bell, S.M.; Burgess, T.; Lee, J.; Blackburn, D.J.; Allen, S.P.; Mortiboys, H. Peripheral Glycolysis in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8924.
- Lu, J.; Qian, J.; Xu, Z.; Yin, S.; Zhou, L.; Zheng, S.; Zhang, W. Emerging Roles of Liquid-Liquid Phase Separation in Cancer: From Protein Aggregation to Immune-Associated Signaling. Front. Cell Dev. Biol. 2021, 9, 631486.
- Petronilho, E.C.; Pedrote, M.M.; Marques, M.A.; Passos, Y.M.; Mota, M.F.; Jakobus, B.; de Sousa, G.D.S.; Pereira da Costa, F.; Felix, A.L.; Ferretti, G.D.S.; et al. Phase Separation of p53 Precedes Aggregation and Is Affected by Oncogenic Mutations and Ligands. Chem. Sci. 2021, 12, 7334–7349.
- Gargini, R.; Segura-Collar, B.; Sánchez-Gómez, P. Novel Functions of the Neurodegenerative-Related Gene Tau in Cancer. Front. Aging Neurosci. 2019, 11, 231.
- Watkins, K.P.; Williams-Carrier, R.; Chotewutmontri, P.; Friso, G.; Teubner, M.; Belcher, S.; Ruwe, H.; Schmitz-Linneweber, C.; van Wijk, K.J.; Barkan, A. Exploring the Proteome Associated with the mRNA Encoding the D1 Reaction Center Protein of Photosystem II in Plant Chloroplasts. Plant J. 2020, 102, 369–382.
- Zoschke, R.; Bock, R. Chloroplast Translation: Structural and Functional Organization, Operational Control, and Regulation. Plant Cell 2018, 30, 745–770.
- Gawroński, P.; Enroth, C.; Kindgren, P.; Marquardt, S.; Karpiński, S.; Leister, D.; Jensen, P.E.; Vinther, J.; Scharff, L.B. Light-Dependent Translation Change of Arabidopsis psbA Correlates with RNA Structure Alterations at the Translation Initiation Region. Cells 2021, 10, 322.
- Preiss, S.; Schrader, S.; Johanningmeier, U. Rapid, ATP-Dependent Degradation of a Truncated D1 Protein in the Chloroplast. Eur. J. Biochem. 2001, 268, 4562–4569.
- Pévet, P. Melatonin in Animal Models. Dialogues Clin. Neurosci. 2003, 5, 343–352.
- Zheng, X.; Tan, D.X.; Allan, A.C.; Zuo, B.; Zhao, Y.; Reiter, R.J.; Wang, L.; Wang, Z.; Guo, Y.; Zhou, J.; et al. Chloroplastic Biosynthesis of Melatonin and Its Involvement in Protection of Plants from Salt Stress. Sci. Rep. 2017, 7, 41236.
- Hwang, O.J.; Kang, K.; Back, K. Effects of Light Quality and Phytochrome Form on Melatonin Biosynthesis in Rice. Biomolecules 2020, 10, 523.
- Zhou, X.; Zhao, H.; Cao, K.; Hu, L.; Du, T.; Baluška, F.; Zou, Z. Beneficial Roles of Melatonin on Redox Regulation of Photosynthetic Electron Transport and Synthesis of D1 Protein in Tomato Seedlings under Salt Stress. Front. Plant Sci. 2016, 7, 1823.