Molecular mechanisms underlying neuropsychiatric and neurodegenerative diseases are insufficiently elucidated. A detailed understanding of these mechanisms may help to further improve medical intervention. Recently, intellectual abilities, creativity, and amnesia have been associated with neuroplastin, a cell recognition glycoprotein of the immunoglobulin superfamily that participates in synapse formation and function and calcium signaling. Data from animal models suggest a role for neuroplastin in pathways affected in neuropsychiatric and neurodegenerative diseases. Neuroplastin loss or disruption of molecular pathways related to neuronal processes has been linked to various neurological diseases, including dementia, schizophrenia, and Alzheimer’s disease
Note: The entry will be online only after author check and submit it.
1. Introduction
The prevalence of mental disorders, including autism spectrum disorder (ASD) and schizophrenia (SZ)
[1][2][3][1,2,3], related to neurodevelopmental deficits and neurodegenerative diseases, is predicted to increase in future decades because of a growing and ageing world population. In addition to its severe effects on cognitive and social communication, its economic burden is a major challenge for patients and for economies at an international level
[4][5][6][7][4,5,6,7]. To understand the pathogenesis of neuropsychiatric disorders, it is important to consider that both genetic and environmental factors can act separately or in combination to play crucial roles in these diseases. Based on genome-wide association studies (GWAS) with large groups of patients, potential mechanisms underlying different psychiatric disorders can be elucidated and directly investigated. As GWAS and next generation sequencing (NGS) have rapidly developed recently, many gene loci have been associated with different neuropsychiatric disorders
[8]. For instance, numerous genetic variants have been associated with SZ
[9][10][11][12][13][9,10,11,12,13] and ASD
[14][15][16][17][14,15,16,17]. Furthermore, a significant genetic correlation exists between ASD and SZ
[17][18][17,18]. In combination with transgenic mouse models targeting the potential risk genes, the association of different psychiatric disorders has been confirmed for explicit genes such as the SHANK genes
[19][20][21][22][23][19,20,21,22,23]. Furthermore, gene variants leading to synaptic dysfunction play a critical role as causal factors for these psychiatric disorders
[24].
The abnormal expression or function of several proteins can affect synaptic transmission and further impair network activities, such as the excitatory and inhibitory balance, contributing to different neuropsychiatric or neurodegenerative diseases
[25][26][25,26]. Therefore, numerous efforts have focused on exploring interventions with identified pathogenic mechanisms, although many attempts did not achieve the desired clinical outcome
[27][28][29][30][31][27,28,29,30,31]. As psychiatric disorders frequently exhibit overlapping symptoms, such as the association of cognitive impairment with ASD and SZ
[32], it is essential to understand their underlying cellular mechanisms.
In this review, we will focus on recent studies of the cell recognition molecule neuroplastin (Np) and its gene (Nptn/NPTN) in relation to psychiatric and neurodegenerative diseases (Figure 1). We propose that targeting neuroplastin may make it possible to reverse network dysfunctions and contribute to ameliorating the onset and progression of neuropsychiatric diseases.
Figure 1. Schematic illustration of neuroplastin Np55/65 as a central component related to neuropsychiatric and neurodegenerative diseases as well as in other diseases associated with neuroplastin malfunctions.
2. Neuroplastin in Neurological Diseases
A particular syndrome has not yet been attributed to neuroplastin malfunction. However, the observed functions in mouse models and the expression, structure, and interaction partners of neuroplastin, indicate that the impairment of neuroplastin function in humans may result in deleterious consequences for the nervous system. Here, we will review the evidence for the contribution of neuroplastin to neurological pathologies.
2.1. Schizophrenia (SZ) and Autism Spectrum Disorder (ASD)
SZ and ASD manifest as distinct neurodevelopmental diseases. ASD frequently presents in childhood, whereas SZ manifests later in young adults. For both disorders, a heritable genetic contribution was observed, but explicit monogenetic causes have not been identified. Furthermore, a significant association between ASD and SZ was detected
[33][101]. Strikingly, many gene loci related to synaptic function were identified as contributing to both SZ and ASD, suggesting that pathological malfunctions of synapses or synaptopathies may be causal (for review see:
[34][102]). In addition, these two diseases frequently co-occur with attention deficit hyperactivity (ADHD) and bipolar disorder (BD); this is likely resulting from a developmental synaptopathy
[35][36][103,104]. Brain images from ASD children have shown increased brain size and weight
[37][105] affecting axons and synaptic density
[38][106], which indicate an acceleration of brain development and more synaptic connections. A lack of adolescent synaptic pruning was observed in ASD patients
[39][107], which may account for the dysfunction of brain circuits in ASD
[40][108].
Unlike ASD, which shows an increase in brain growth in all brain regions except occipital grey matter
[41][109], loss of grey matter in SZ was observed
[42][110]. Excessive synaptic pruning in prefrontal cortical synapses was found in SZ neuropathology
[43][111], which indicated reduced synapses and further impairments of the brain circuitry and cognitive functions
[44][45][112,113].
2.1.1. Neuroplastin Relation to Schizophrenia
In rat models displaying schizophrenia-like symptoms after injection of the two different psychostimulants methamphetamine (MAP) or phencyclidine (PCP), neuroplastin was significantly up-regulated
[46][114]. MAP is a dopamine transporter inhibitor that causes a positive symptom, clinically similar to paranoid schizophrenia in an acute phase
[47][115]. PCP is an NMDA receptor antagonist, which induces both negative and positive schizophrenia-like symptomatology
[48][116]. Subsequent genetic studies of patients with schizophrenia identified three single-nucleotide polymorphisms (SNPs) in the 5′-upstream region of
NPTN that were strongly correlated to schizophrenia
[49][44].
Pre-pulse inhibition (PPI) of the startle response is often considered as a characteristic in the diagnosis of schizophrenia
[50][117]. In
Nptn-deficient mice, PPI is severely impaired
[51][50], although this could simply result from the profound hearing deficit of these mice
[52][43], rather than processing deficits. Nevertheless, the significant reduction in paired-pulse facilitation in the auditory cortex of
Nptn-deficient mice suggests altered cortical synaptic transmission
[52][43]. In addition, the PPI deficit in heterozygous
Nptn-deficient mice
[51][50] points to potential central alterations as contributors to the phenotype. As detailed above, the neuroplastin interaction partners AMPA receptor subunit GluA1 and TRAF6 have also been associated with schizophrenia.
2.1.2. Autism Spectrum Disorder (ASD)
Some patients suffering from the heterogeneous 15q24 microdeletion syndrome display ASD and attention deficit hyperactivity disorder (ADHD), in addition to various other deficits
[53][54][118,119].
NPTN is located at cytogenetic band 15q24.1 and it is deleted or duplicated in some 15q24 microdeletion syndrome patients
[53][54][118,119]. Furthermore, PMCA2 was identified by GWAS studies to be associated with ASD
[55][120]. A study which included 717 children associated the cortical morphology, such as cortical thickness and surface area, with autistic traits
[56][121]. Interestingly, a single-nucleotide polymorphism in
NPTN was found to be associated with cortical thickness
[57][49]. Furthermore, the paths to ASDs may involve unbalanced excitatory–inhibitory synaptic transmission and abnormal synaptogenesis
[58][122]. Several studies have observed an imbalance of excitatory-inhibitory transmission and altered synaptogenesis in different
Nptn-deficient mice
[59][60][61][62][63][40,51,59,60,123]. In addition, neuroplastin-deficient mice displayed altered social interactions avoiding unfamiliar mice
[51][50]. In conclusion, genetic association studies suggest a link of neuroplastin to autism spectrum disorder, but the role of
NPTN in ASD still needs to be specifically addressed. It remains to be seen whether a direct malfunction or loss of neuroplastin, rather than an indirect effect, e.g., via PMCAs, contributes to ASD.
2.2. Depression and Anxiety Disorder
Depression and anxiety are the most common mental disorders in society today, and both frequently co-occur in patients
[64][65][124,125]. Etiological factors related to depression and anxiety disorder could be linked to childhood trauma, environmental adversity, as well as stressful life events
[66][126]. Furthermore, several genes have been associated with depression and anxiety, among them
5-HTT,
NPSR1, and
RGS2 [66][126]. Genetic inactivation of
Nptn results in elevated corticosterone levels and increased depressive-like behavior, but reduced anxiety-related behaviors in mice
[51][50]. Mice that lacked only Np65 displayed the opposite phenotype, with reduced depressive-like behavior and increased anxiety
[61][59]. In addition, the neuroplastin binding partners GluA1 and GABA
A receptor are associated with anxiety disorder and depression.
2.3. Alzheimer’s (AD) Disease
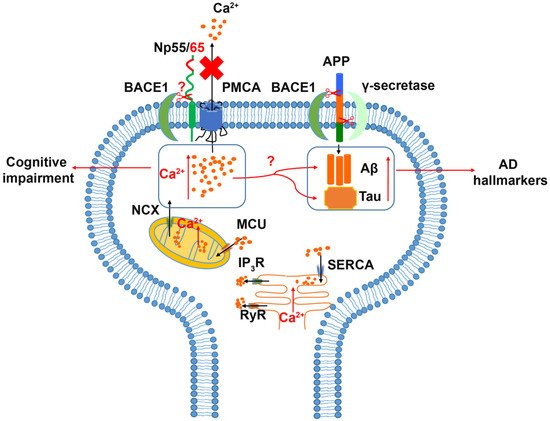
An alteration of neuroplastin expression in AD patients was reported recently
[67][58]. In the early phase of confirmed AD neuropathology, neuroplastin was significantly upregulated in the hippocampus (dentate gyrus, CA2/3 region, and subiculum) without changes in neuron number or tissue volume. Interestingly, patients experiencing a longer duration of AD disease (5–7 years) showed a decreased expression level of neuroplastin compared to patients with a short duration AD (≤4 years), which may indicate a role of neuroplastin in the early phase of AD. The analysis of neuropathological amyloid plaques and neurofibrillary tangles (NFT) showed a negative correlation between neuroplastin expression level and the number of amyloid plaques in the CA1 area and a weak negative correlation between neuroplastin and NFT in CA1, CA2/3, and subiculum. In human brain, both
NPTN and
PMCAs exhibit similar expression patterns at the transcriptomic level
[68][127]. In comparison to the aging brain, the expression and activity of PMCA in AD patients were reduced, and the AD hallmarks tau and Aβ showed a negative impact on PMCA activity, which may indicate an altered Ca
2+ homeostasis in the AD brain
[69][70][71][96,128,129]. Alternatively, Ca
2+ dys-homeostasis could promote the accumulation of Aβ and phosphorylated tau protein, which result in the neuropathy and brain function deficits in AD patients
[70][71][72][128,129,130]. Aβ is produced by the β- and γ-secretase cleavages of the amyloid precursor protein (APP)
[73][131]. The principal β-secretase for generation of Aβ in vivo is the β-site APP cleaving enzyme 1 (BACE1)
[73][74][131,132]. The use of BACE1-specific inhibitors has been proposed as a potential intervention in AD
[75][133]; however, this approach must be regarded carefully, as BACE1 cleaves numerous substrates. Interestingly, both neuroplastin and basigin were identified as potential BACE1 substrates
[76][77][134,135]. Although further studies on the cleavage of Np by BACE1 were not conducted, an attractive hypothesis is that increased neuroplastin cleavage by BACE1 could result in the cognitive deficits observed in AD (
Figure 2).
Figure 2. Relation of Neuroplastin to Alzheimer’s disease. The Amyloid Precursor Protein (APP) is cleaved aberrantly by β-secretase (BACE1) and γ-secretase resulting in Aβ. Aβ and tau are considered hallmarks of Alzheimer’s disease. Increased intracellular Ca2+ concentrations are associated with cognitive impairment and increases in Aβ and tau. Intracellular Ca2+ can be deposited into or released from mitochondria and ER as intracellular calcium stores. Energy driven extrusion of Ca2+ is mediated by PMCAs. In the absence of neuroplastin, PMCA levels are reduced and intracellular Ca2+ is increased. The hypothetical cleavage of neuroplastin by BACE1 may result in loss of PMCAs and elevated Ca2+ levels.
2.4. Cognition, Antero- and Retrograde Amnesia
A large-scale genetic association study in adolescents associated
NPTN with cortical thickness and intellectual ability
[57][49], suggesting a role for NPTN in cognition and learning and memory. Furthermore,
NPTN variants were recently related to creativity
[78][136].
In recent years, we have addressed the role of neuroplastin in learning and memory using several mouse mutants. When neuroplastin was missing from only glutamatergic neurons, achieved using an
Emx1-promoter driven Cre-recombinase, associative learning was slightly improved. However, the continuity of task execution was affected, suggesting altered striatum-dependent decision making
[79][35]. The complete loss of neuroplastin expression resulted in a complex phenotype, which included the inability to learn associative tasks
[51][50]. The comparison of
Nptn-ablation in glutamatergic versus all neurons suggests a particular role of neuroplastin, expressed by gabaergic interneurons for associative learning. When neuroplastin expression was specifically ablated in all types of neurons after a normal development, again, the anterograde memory was not formed for associative tasks
[51][50]. Furthermore, when the associative tasks were first acquired perfectly before neuroplastin ablation, the induced loss of neuroplastin resulted in specific retrograde amnesia for these associative memories but not for spatial memories
[51][50]. Interestingly, the β-blocker propranolol has retrograde amnestic side effects. Therefore, propranolol is applied as off-label use for the treatment of intrusive thoughts associated with post-traumatic stress disorder (PTSD) (for a review, see:
[80][137]). Propranolol also acts as an inhibitor of the PMCAs
[81][138], and thus its amnestic effects may be related to PMCA inhibition. In vivo, the absence of neuroplastin leads to dramatically reduced PMCA levels
[79][51][35,50], suggesting that the neuroplastin-PMCA assembly may be critical for associative learning and memory. If this hypothesis can be substantiated, it may provide an opportunity to address PTSD and other psychiatric conditions involving intrusive thoughts.
2.5. Other Diseases Related to Neuroplastin
2.5.1. Deafness
Deafness resulting from the loss of neuroplastin function has been studied using
Nptn-deficient and neuroplastin mutant mice
[52][60][82][43,51,52]. It was proposed that Np55 expression by outer hair cells is required for cochlear amplification
[82][52], and Carrott et al.
[60][51] proposed Np65-driven synaptogenesis by inner hair cells as necessary for hearing. Recently, we showed that neuroplastin expression is essential for hearing during the development of the hearing system, and also for the maintenance of hearing capabilities in adults throughout their life
[52][43]. Neuroplastin is required for PMCA2 targeting and Ca
2+ extrusion in cochlear hair cells
[52][43]. Interestingly, PMCA2 loss of function mutations result in deafness in mice and human
[83][139], verifying that the interaction of neuroplastin with PMCA is decisive for Ca
2+ extrusion.
2.5.2. Cancer
The first evidence linking
NPTN to cancer came from a bioinformatic analysis showing that
NPTN was one of 166 genes with an altered expression in colorectal adenomatous polyps
[84][140]. In a study screening for potential tumor antigens from breast cancer patients, neuroplastin was identified and showed strongly increased immunoreactivity in invasive carcinoma tissues
[85][141]. Moreover, over-expression of neuroplastin in a breast-cancer-derived cell line strongly increased tumor growth and angiogenesis, as well as the production of vascular endothelia growth factor (VEGF), suggesting an angiogenic mechanism regulated by VEGF in the aberrant neuroplastin-expressing tumors
[85][141]. Furthermore, the role of neuroplastin and its interaction with S100A8/A9, resulting in the activation of a cascade for lung cancer disseminative progression and aggressive development, has been proposed
[86][87][142,143].
2.5.3. Various Diseases
Not surprisingly, the widespread expression of neuroplastin in nearly all organs may result in the discovery of further pathological conditions influenced by neuroplastin, e.g., within the immune system
[88][55] or in heart disease
[89][144].