Fracture-healing is a complex multi-stage process that usually progresses flawlessly, resulting in restoration of bone architecture and function. Regrettably, however, a considerable number of fractures fail to heal, resulting in delayed unions or non-unions. This may significantly impact several aspects of a patient’s life.
- fracture-healing
- bone remodeling
- bone ossification
- bone marrow adiposity
- skeletal stem cells
1. Introduction
Fracture is a very frequent cause of morbidity with one third of males and one half of females being affected globally, at least once in their lifetime. Even though most fractures recover uneventfully, 2–11% of fractures, depending on which long bone has been traumatized, fail to unite, resulting in non-unions [1]. Importantly, this may strongly affect many aspects of the patient’s life leading to financial, family-related, and psychological issues. Indeed, it has been reported that the average direct cost of long bone non-union treatment is USD 11,333 (USA), USD 11,800 (Canada), and GBP 29,204 (UK) [2]. Not surprisingly, the socioeconomic burden imposed on the healthcare system by fractures has pushed the scientific community towards a synchronized effort to delve deeper and shed light upon the mechanisms of fracture repair to develop novel more effective therapeutic interventions for the successful management of bone injuries [3][4][3,4] For this to be achieved, a multidisciplinary approach employing different scientific specializations (i.e., medicine, biology, biomechanics, material sciences) is vital.
Fractures result in disruption of bone continuity, which in turn leads to changes in bone microenvironment and homeostasis. The process of fracture-healing aims at bridging the gap between the bone parts and at ensuring that injured bone will regain its pre-fracture structural and functional characteristics. To achieve this, a series of cellular and molecular events that recapitulate normal bone development take place. Two major types of fracture-healing processes have been recognized: (a) the primary (direct) healing, during which skeletal stem cells (SSC) directly differentiate into osteoblasts mirroring intramembranous bone formation; and (b) the secondary (indirect) healing that recapitulates mainly endochondral bone formation, since SSC transform to chondroblasts, which are gradually replaced by bone-forming osteoblasts [5].
2. The Catabolic Phase of Fracture-Healing
The catabolic phase of fracture-healing is accomplished by the bone-resorbing cells of the skeleton, the osteoclasts (OCL) ( Figure 1 ). OCL are “surface cells” and as such, they must tightly attach bone surfaces, through specialized integrin (mainly ανβ3) adhesions, to exert their function. Cell membrane infoldings, rich in actin filaments, called ruffled border, are responsible for the attachment of OCL to bone surfaces and mark the area to be resorbed. Acid-secreting H+-ATPases [6][41] assist acid secretion to the resorption (Howship’s) lacunae, which is further enhanced by chloride-proton exchange and chloride channels [7][8][42,43]. OCL secret specific acid proteinases, namely cathepsins K and matrix metalloproteases (MMP)-9 and -13 that degrade collagen and other ECM constituents. Bone debris is removed primarily by vacuolar trafficking [9][14].
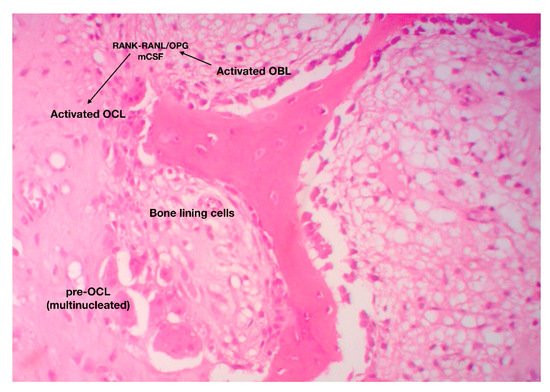
The function of OCL is under the direct and/or indirect control of endocrine/systemic (calcitonin, PTH) and local (paracrine) factors. It has been previously mentioned that OCL differentiation, proliferation and maturation relies upon the RANK-RANKL/OPG axis and m-CSF-dependent pathways [10][44].
OCL participates in several steps of fracture-healing. More specifically, during the early, inflammatory phase, osteoclastic activity is peaking due to the need for removal of excess debris and extracellular matrix from the wounded area. This process is mediated by neutrophil-derived pro-inflammatory cytokines such as TNF-a, IL-6, CCl-2, which recruit macrophages and activate signaling pathways that promote osteoclastic activity [10][44]. During indirect fracture-healing, OCL remove calcified cartilage from the injured area paving the way towards bone synthesis. Some authors use the term “chondroclasts” to describe these cartilage-destroying multinucleate giant cells; nonetheless it is now widely accepted that these cells share the same morphological and molecular feature with their bone-resorbing counterparts, the OCL. Finally, during the catabolic phase of fracture repair, OCL degrade and remove the excess of bony (hard) callus, reshape bone and eventually restore the pre-fracture architectural and functional features of the bones. Even though the anabolic phase of fracture-healing is completed in about 10–12 days, the catabolic phase lasts from 1 month to 2 years [11][45]. Interestingly, several studies and animal models have shown that inhibition of OCL has minor effect on cartilage but major effect on bone callus remodeling [12][46].
As with anabolic phase, the catabolic phase of fracture repair is also strongly influenced by mechanical stimulation. Indeed, reduced mechanical stimulation results in enhanced production of pro-inflammatory cytokines such as IL-6, TNF-α by osteoblasts, directly promoting osteoclast differentiation and maturation. In addition, low levels of mechanical loading lead in over-production of m-CSF and increase the RANKL/OPG ratio, favoring osteoclast activation and hence bone resorption [13][12].
3. The Role of Bone Marrow Fat in Fracture-Healing Process
Despite the prevailing view that adipose tissue is just a metabolically blunt, space occupying tissue, it is now well established that it exhibits significant metabolic activity and participates in vital physiological functions of the body, including response to endocrine and inflammatory stimuli. According to the classical concept, there are two types of adipose tissue: the white (WAT) and brown adipose tissue (BAT) that have distinct origin and different morphology and function.
From a micromorphological perspective, white adipocytes are spherical with centrally situated endocytoplasmic lipid droplet that pushes nucleus towards cell periphery. WAT regulates the balance between energy/nutritional availability and nutritional needs of the body, acting as a storehouse of excess calories. Moreover, it protects non-fatty tissues from toxic accumulation of nutrients [14][47]. WAT can communicate with other organs via endocrine mechanisms mediated by a variety fat-specific cytokines called lipokines [15][48].
In recent years, a third category of adipose tissue that shares common features with both WAT and BAT, has been described. For obvious reasons, it is called “brite” or “beige” adipose tissue (BrAT). Brite adipocytes originate from Myf5- precursors, similarly to WAT; nevertheless, they are multivacuolated cells, rich in UCP1 expressing mitochondria. For this reason, it is believed that BrAT are morphologically and functionally closer to BAT. Notably, BrAT has greater thermogenic capacity than BAT, resulting in higher UCP1 levels [16][51].
Low-grade chronic inflammation has also significant impact on the regulation of osteoclast function. More specifically, activated T-lymphocytes promote the synthesis of RANKL and prevent the production of OPG by osteoblasts, resulting in osteoclastic activation and maturation [17][54]. Notably, in skeleton, bone marrow adipocytes can induce osteoclastogenesis via RANK-RANKL-dependent and -independent mechanisms [18][55]. Additionally, the inflammatory ecosystem induces the synthesis of MCP1 (monocyte chemoattractant protein 1), a cytokine involved in the activation of several osteoclast-related signaling pathways, such as the JAK-STAT [18][55]. The effect of bone marrow BAT and WAT on bone metabolism has not been thoroughly investigated. Nonetheless, ample evidence indicates that the presence of BrAT and/or the conversion of WAT to BAT, have an anabolic effect on bones, which can be attributed, at least in part, to cytokines such as IL-1 and to modifications of sympathetic nervous system [19][56]. Very recent studies have shown that HDL deficiency is associated with reduced bone mass and decreased expression of specific BAT-related genes, suggesting a mechanistic link between HDL, bone marrow fat and bone metabolism [20][21][57,58].
4. Adipogenesis, Osteogenesis and Fracture-Healing
Noteworthy, mounting body of evidence over the past few years has pointed out that bone marrow adipose tissue greatly influences bone homeostasis and function. As a matter of fact, lipoblasts and osteoblasts represent the two sides of the same coin and thus, increased levels of one cell type are associated with decreased levels of the other. In line with that, increased marrow adiposity is followed by reduced bone mass, a phenomenon that is governed by a series of complex cellular and molecular events. LBL secrete factors such as chemerin, resistin, visfatin, leptin, adiponectin and omentin-1 that affect SSC program enhancing adipogenesis, while impeding osteoblastogenesis [22][59]. Others suggest that these molecules exert a direct action on hematopoietic stem cells HSC promoting osteoclastogenesis. In the same vein, bone marrow adipocytes synthesize RANKL, further contributing to OCL differentiation and maturation [22][59]. The augmented levels of inflammatory cytokines and free fatty acid from BM adipocytes enhance the lipogenic shift of SSC and osteoclastogenesis, affecting the susceptibility of bones to fractures, and their ability for fracture repair.
The tight connection between bone homeostasis and bone marrow adiposity is further reinforced by recent studies having investigated the role of HDL in the configuration of BM microenvironment. These studies have shown that HDL deficiency results in elevated BM adiposity and reduced bone mass, an effect that is mainly attributed to suppressed osteoblastogenesis, since osteoclast number and function remain unaffected [9][21][14,58].
Since bone marrow adiposity affects bone marrow microenvironment and skeletal biology, it could be speculated that it also affects the susceptibility of bone to fractures as well as its ability for uneventful healing. In fact, there are several research findings that support this speculation. Elevated BM adiposity is associated with the development of a pro-inflammatory microenvironment, which as mentioned previously, promotes lipo- and osteoclastogenesis resulting in reduced bone mass and hence increased susceptibility of bones to fractures. In addition, pathological conditions (i.e., diabetes type II, ageing, osteoporosis, HDL deficiency, metabolic syndrome) characterized by increased marrow adiposity and by a switch towards WAT, further increase the pro-inflammatory milieu of BM and hence, the capability of bone to recover from fractures, for all the reasons that have been addressed earlier in this paper.
Unraveling the molecular interface between bone and fat, it is likely to lead to the development of novel pharmaceutical agents (such as HDL-directed molecules) that could modify bone marrow microenvironment and accelerate fracture-healing progress.
References
- Nandra, R.; Grover, L.; Porter, K. Fracture non-union epidemiology and treatment. Trauma 2016, 18, 3–11, doi:10.1177/1460408615591625.
- Hak, D.J.; Fitzpatrick, D.; Bishop, J.A.; Marsh, J.L.; Tilp, S.; Schnettler, R.; Simpson, H.; Alt, V. Delayed union and nonunions: Epidemiology, clinical issues, and financial aspects. Injury 2014, 45, S3–S7, doi:10.1016/j.injury.2014.04.002.
- Melton, L.J.; Gabriel, S.E.; Crowson, C.S.; Tosteson, A.N.A.; Johnell, O.; Kanis, J.A. Cost-equivalence of different osteoporotic fractures. Osteoporos. Int. 2003, 14, 383–388, doi:10.1007/s00198-003-1385-4.
- Johnell, O.; Kanis, J. Epidemiology of osteoporotic fractures. Osteoporos. Int. 2005, 16, S3–S7, doi:10.1007/s00198-004-1702-6.
- Collignon, A.-M.; Lesieur, J.; Vacher, C.; Chaussain, C.; Rochefort, G.Y. Strategies Developed to Induce, Direct, and Potentiate Bone Healing. Front. Physiol. 2017, 8, 927, doi:10.3389/fphys.2017.00927.
- Robey, P. “Mesenchymal stem cells”: Fact or fiction, and implications in their therapeutic use. F1000Research 2017, 6, doi:10.12688/f1000research.10955.1.
- Serowoky, M.A.; Arata, C.E.; Crump, J.G.; Mariani, F.V. Skeletal stem cells: Insights into maintaining and regenerating the skeleton. Development 2020, 147, dev179325, doi:10.1242/dev.179325.
- Chan, C.K.; Seo, E.Y.; Chen, J.Y.; Lo, D.; McArdle, A.; Sinha, R.; Tevlin, R.; Seita, J.; Vincent-Tompkins, J.; Wearda, T.; et al. Identification and specification of the mouse skeletal stem cell. Cell 2015, 160, 285–298, doi:10.1016/j.cell.2014.12.002.
- Sutures Possess Strong Regenerative Capacity for Calvarial Bone Injury. Stem Cells Dev. 2016, 25, 1801–1807, doi:10.1089/scd.2016.0211.
- Park, D.; Spencer, J.A.; Koh, B.I.; Kobayashi, T.; Fujisaki, J.; Clemens, T.L.; Lin, C.P.; Kronenberg, H.M.; Scadden, D.T. Endogenous bone marrow MSCs are dynamic, fate-restricted participants in bone maintenance and regeneration. Cell Stem Cell 2012, 10, 259–272, doi:10.1016/j.stem.2012.02.003.
- Wang, T.; Zhang, X.; Bikle, D.D. Osteogenic Differentiation of Periosteal Cells during Fracture Healing. J. Cell. Physiol. 2017, 232, 913–921, doi:10.1002/jcp.25641.
- Papachristou, D.J.; Papachroni, K.K.; Basdra, E.K.; Papavassiliou, A.G. Signaling networks and transcription factors regulating mechanotransduction in bone. BioEssays 2009, 31, 794–804, doi:10.1002/bies.200800223.
- Blair, H.C.; Zaidi, M.; Huang, C.L.-H.; Sun, L. The Developmental Basis of Skeletal Cell Differentiation and the Molecular Basis of Major Skeletal Defects. Biol. Rev. 2008, 83, 401–415, doi:10.1111/j.1469-185X.2008.00048.x.
- de Gorter, D.J.J.; ten Dijke, P. Signal Transduction Cascades Controlling Osteoblast Differentiation. In Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism; Wiley Online Books: UK, 2013; pp. 15-24.
- Crockett, J.C.; Mellis, D.J.; Scott, D.I.; Helfrich, M.H. New knowledge on critical osteoclast formation and activation pathways from study of rare genetic diseases of osteoclasts: Focus on the RANK/RANKL axis. Osteoporos. Int. 2011, 22, 1–20, doi:10.1007/s00198-010-1272-8.
- Zhang, J.; Niu, C.; Ye, L.; Huang, H.; He, X.; Tong, W.-G.; Ross, J.; Haug, J.; Johnson, T.; Feng, J.Q.; et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature 2003, 425, 836–841, doi:10.1038/nature02041.
- Bonewald, L.F. The amazing osteocyte. J. Bone Miner. Res. 2011, 26, 229–238, doi:10.1002/jbmr.320.
- Jacobs, C.R.; Temiyasathit, S.; Castillo, A.B. Osteocyte mechanobiology and pericellular mechanics. Annu. Rev. Biomed. Eng. 2010, 12, 369–400, doi:10.1146/annurev-bioeng-070909-105302.
- Tatsumi, S.; Ishii, K.; Amizuka, N.; Li, M.; Kobayashi, T.; Kohno, K.; Ito, M.; Takeshita, S.; Ikeda, K. Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab. 2007, 5, 464–475, doi:10.1016/j.cmet.2007.05.001.
- Rochefort, G.Y.; Pallu, S.; Benhamou, C.L. Osteocyte: The unrecognized side of bone tissue. Osteoporos. Int. 2010, 21, 1457–1469, doi:10.1007/s00198-010-1194-5.
- Martin, T.J.; Sims, N.A. RANKL/OPG; Critical role in bone physiology. Rev. Endocr. Metab. Disord. 2015, 16, 131–139, doi:10.1007/s11154-014-9308-6.
- Marsell, R.; Einhorn, T.A. The biology of fracture healing. Injury 2011, 42, 551–555, doi:10.1016/j.injury.2011.03.031.
- Jacobsen, K.A.; Al-Aql, Z.S.; Wan, C.; Fitch, J.L.; Stapleton, S.N.; Mason, Z.D.; Cole, R.M.; Gilbert, S.R.; Clemens, T.L.; Morgan, E.F.; et al. Bone Formation During Distraction Osteogenesis Is Dependent on Both VEGFR1 and VEGFR2 Signaling. J. Bone Miner. Res. 2008, 23, 596–609, doi:10.1359/jbmr.080103.
- Riddle, R.C.; Khatri, R.; Schipani, E.; Clemens, T.L. Role of hypoxia-inducible factor-1α in angiogenic–osteogenic coupling. J. Mol. Med. 2009, 87, 583–590, doi:10.1007/s00109-009-0477-9.
- Ceradini, D.J.; Kulkarni, A.R.; Callaghan, M.J.; Tepper, O.M.; Bastidas, N.; Kleinman, M.E.; Capla, J.M.; Galiano, R.D.; Levine, J.P.; Gurtner, G.C. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat. Med. 2004, 10, 858–864, doi:10.1038/nm1075.
- Kolar, P.; Schmidt-Bleek, K.; Schell, H.; Gaber, T.; Toben, D.; Schmidmaier, G.; Perka, C.; Buttgereit, F.; Duda, G.N. The early fracture hematoma and its potential role in fracture healing. Tissue Eng. Part B Rev. 2010, 16, 427–434, doi:10.1089/ten.TEB.2009.0687.
- Yuasa, M.; Mignemi, N.A.; Nyman, J.S.; Duvall, C.L.; Schwartz, H.S.; Okawa, A.; Yoshii, T.; Bhattacharjee, G.; Zhao, C.; Bible, J.E.; et al. Fibrinolysis is essential for fracture repair and prevention of heterotopic ossification. J. Clin. Investig. 2015, 125, 3117–3131, doi:10.1172/JCI80313.
- Kovtun, A.; Bergdolt, S.; Wiegner, R.; Radermacher, P.; Huber-Lang, M.; Ignatius, A. The crucial role of neutrophil granulocytes in bone fracture healing. Eur. Cell Mater. 2016, 32, 152–162, doi:10.22203/ecm.v032a10.
- Kon, T.; Cho, T.-J.; Aizawa, T.; Yamazaki, M.; Nooh, N.; Graves, D.; Gerstenfeld, L.C.; Einhorn, T.A. Expression of Osteoprotegerin, Receptor Activator of NF-κB Ligand (Osteoprotegerin Ligand) and Related Proinflammatory Cytokines During Fracture Healing. J. Bone Miner. Res. 2001, 16, 1004–1014, doi:10.1359/jbmr.2001.16.6.1004.
- Pettit, A.R.; Chang, M.K.; Hume, D.A.; Raggatt, L.-J. Osteal macrophages: A new twist on coupling during bone dynamics. Bone 2008, 43, 976–982, doi:10.1016/j.bone.2008.08.128.
- Yamaguchi, A.; Sakamoto, K.; Minamizato, T.; Katsube, K.; Nakanishi, S. Regulation of osteoblast differentiation mediated by BMP, Notch, and CCN3/NOV. Jpn. Dent. Sci. Rev. 2008, 44, 48–56, doi:10.1016/j.jdsr.2007.11.003.
- Liang, H.P.H.; Xu, J.; Xue, M.; Jackson, C. Matrix metalloproteinases in bone development and pathology: Current knowledge and potential clinical utility. Met. Med. 2016, 3, 93–102, doi:10.2147/mnm.s92187.
- Sinder, B.P.; Pettit, A.R.; McCauley, L.K. Macrophages: Their Emerging Roles in Bone. J. Bone Miner. Res. 2015, 30, 2140–2149, doi:10.1002/jbmr.2735.
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795, doi:10.1172/jci59643.
- Mailhot, G.; Yang, M.; Mason-Savas, A.; MacKay, C.A.; Leav, I.; Odgren, P.R. BMP-5 expression increases during chondrocyte differentiation in vivo and in vitro and promotes proliferation and cartilage matrix synthesis in primary chondrocyte cultures. J. Cell. Physiol. 2008, 214, 56–64, doi:10.1002/jcp.21164.
- Ye, F.; Xu, H.; Yin, H.; Zhao, X.; Li, D.; Zhu, Q.; Wang, Y. The role of BMP6 in the proliferation and differentiation of chicken cartilage cells. PLoS ONE 2019, 14, e0204384, doi:10.1371/journal.pone.0204384.
- Rosati, R.; Horan, G.S.B.; Pinero, G.J.; Garofalo, S.; Keene, D.R.; Horton, W.A.; Vuorio, E.; de Crombrugghe, B.; Behringer, R.R. Normal long bone growth and development in type X collagen-null mice. Nat. Genet. 1994, 8, 129–135, doi:10.1038/ng1094-129.
- Day, T.F.; Guo, X.; Garrett-Beal, L.; Yang, Y. Wnt/β-Catenin Signaling in Mesenchymal Progenitors Controls Osteoblast and Chondrocyte Differentiation during Vertebrate Skeletogenesis. Dev. Cell 2005, 8, 739–750, doi:10.1016/j.devcel.2005.03.016.
- Hill, T.P.; Später, D.; Taketo, M.M.; Birchmeier, W.; Hartmann, C. Canonical Wnt/β-Catenin Signaling Prevents Osteoblasts from Differentiating into Chondrocytes. Dev. Cell 2005, 8, 727–738, doi:10.1016/j.devcel.2005.02.013.
- Hu, D.P.; Ferro, F.; Yang, F.; Taylor, A.J.; Chang, W.; Miclau, T.; Marcucio, R.S.; Bahney, C.S. Cartilage to bone transformation during fracture healing is coordinated by the invading vasculature and induction of the core pluripotency genes. Development 2017, 144, 221–234, doi:10.1242/dev.130807.
- Blair, H.; Teitelbaum, S.; Ghiselli, R.; Gluck, S. Osteoclastic bone resorption by a polarized vacuolar proton pump. Science 1989, 245, 855–857, doi:10.1126/science.2528207.
- Palagano, E.; Blair, H.C.; Pangrazio, A.; Tourkova, I.; Strina, D.; Angius, A.; Cuccuru, G.; Oppo, M.; Uva, P.; Van Hul, W.; et al. Buried in the Middle but Guilty: Intronic Mutations in the TCIRG1 Gene Cause Human Autosomal Recessive Osteopetrosis. J. Bone Miner. Res. 2015, 30, 1814–1821, doi:10.1002/jbmr.2517.
- Schlesinger, P.H.; Blair, H.C.; Teitelbaum, S.L.; Edwards, J.C. Characterization of the Osteoclast Ruffled Border Chloride Channel and Its Role in Bone Resorption *. J. Biol. Chem. 1997, 272, 18636–18643, doi:10.1074/jbc.272.30.18636.
- Lelliott, C.; Vidal-Puig, A.J. Lipotoxicity, an imbalance between lipogenesis de novo and fatty acid oxidation. Int. J. Obes. 2004, 28, S22–S28, doi:10.1038/sj.ijo.0802854.
- Einhorn, T.A.; Gerstenfeld, L.C. Fracture healing: Mechanisms and interventions. Nat. Rev. Rheumatol. 2015, 11, 45–54, doi:10.1038/nrrheum.2014.164.
- Einhorn, T.A. Can an anti-fracture agent heal fractures? Clin. Cases Miner Bone Metab. 2010, 7, 11–14.
- Scherer, P.E. Adipose Tissue. Lipid Storage Compart. Endocr. Organ 2006, 55, 1537–1545, doi:10.2337/db06-0263.
- Cannon, B.; Nedergaard, J. Brown Adipose Tissue: Function and Physiological Significance. Physiol. Rev. 2004, 84, 277–359, doi:10.1152/physrev.00015.2003.
- Gesta, S.; Tseng, Y.-H.; Kahn, C.R. Developmental Origin of Fat: Tracking Obesity to Its Source. Cell 2007, 131, 242–256, doi:10.1016/j.cell.2007.10.004.
- de Jesus, L.A.; Carvalho, S.D.; Ribeiro, M.O.; Schneider, M.; Kim, S.-W.; Harney, J.W.; Larsen, P.R.; Bianco, A.C. The type 2 iodothyronine deiodinase is essential for adaptive thermogenesis in brown adipose tissue. J. Clin. Investig. 2001, 108, 1379–1385, doi:10.1172/JCI13803.
- Wu, J.; Boström, P.; Sparks, L.M.; Ye, L.; Choi, J.H.; Giang, A.-H.; Khandekar, M.; Virtanen, K.A.; Nuutila, P.; Schaart, G.; et al. Beige Adipocytes Are a Distinct Type of Thermogenic Fat Cell in Mouse and Human. Cell 2012, 150, 366–376, doi:10.1016/j.cell.2012.05.016.
- Clevers, H. Wnt/beta-catenin signaling in development and disease. Cell 2006, 127, 469–480, doi:10.1016/j.cell.2006.10.018.
- Kaneki, H.; Guo, R.; Chen, D.; Yao, Z.; Schwarz, E.M.; Zhang, Y.E.; Boyce, B.F.; Xing, L. Tumor Necrosis Factor Promotes Runx2 Degradation through Up-regulation of Smurf1 and Smurf2 in Osteoblasts. J. Biol. Chem. 2006, 281, 4326–4333, doi:10.1074/jbc.M509430200.
- Ma, T.; Miyanishi, K.; Suen, A.; Epstein, N.J.; Tomita, T.; Smith, R.L.; Goodman, S.B. Human interleukin-1-induced murine osteoclastogenesis is dependent on RANKL, but independent of TNF-α. Cytokine 2004, 26, 138–144, doi:10.1016/j.cyto.2004.02.001.
- Redlich, K.; Smolen, J.S. Inflammatory bone loss: Pathogenesis and therapeutic intervention. Nat. Rev. Drug Discov. 2012, 11, 234–250, doi:10.1038/nrd3669.
- Motyl, K.J.; Bishop, K.A.; DeMambro, V.E.; Bornstein, S.A.; Le, P.; Kawai, M.; Lotinun, S.; Horowitz, M.C.; Baron, R.; Bouxsein, M.L.; et al. Altered thermogenesis and impaired bone remodeling in Misty mice. J. Bone Miner. Res. 2013, 28, 1885–1897, doi:10.1002/jbmr.1943.
- Blair, H.C.; Kalyvioti, E.; Papachristou, N.I.; Tourkova, I.L.; Syggelos, S.A.; Deligianni, D.; Orkoula, M.G.; Kontoyannis, C.G.; Karavia, E.A.; Kypreos, K.E.; et al. Apolipoprotein A-1 regulates osteoblast and lipoblast precursor cells in mice. Lab. Investig. 2016, 96, 763–772, doi:10.1038/labinvest.2016.51.
- Papachristou, N.I.; Blair, H.C.; Kypreos, K.E.; Papachristou, D.J. High-density lipoprotein (HDL) metabolism and bone mass. J. Endocrinol. 2017, 233, R95–R107, doi:10.1530/joe-16-0657.
- Muruganandan, S.; Sinal, C.J. The impact of bone marrow adipocytes on osteoblast and osteoclast differentiation. IUBMB Life 2014, 66, 147–155, doi:10.1002/iub.1254.