Maintaining the effects of nematode-trapping fungi (NTF) agents in order to control plant-parasitic nematodes (PPNs) in different ecological environments has been a major challenge in biological control applications. To achieve such an objective, it is important to understand how populations of the biocontrol agent NTF are geographically and ecologically structured.
- short tandem repeat
- genetic differentiation
- unique alleles
- non-random recombination
1. Introduction
Plant-parasitic nematodes (PPNs), especially the root-knot nematodes in the genus Meloidogyne, are widespread pests that cause crop yield losses worth more than US $157 billion worldwide each year [1]. For decades, the control of Meloidogyne spp. has heavily relied on chemical nematicides. However, resistance to chemical nematicides has emerged, and the environmental and human health impacts of nematicide residues are becoming increasingly recognized[2]. Therefore, currently available chemical nematicides are being phased out, and an increasing number of biocontrol agents are being introduced to help control PPNs[3].
At present, about 700 fungal species are known to be capable of attacking living nematodes (juveniles, adults, and eggs). These fungi are taxonomically diverse but traditionally divided into five groups based on the mechanisms by which they attack nematodes: (i) nematode-trapping fungi (NTF), (ii) endoparasitic fungi, (iii) egg- and cyst-parasitic fungi, (iv) toxin-producing fungi, and (v) fungi with special nematode-attacking devices [4]. Among these groups, the broad adaptability and flexible lifestyles of NTFs make them ideal agents for controlling PPNs [4][5][6][7]. NTFs are abundantly distributed in a broad range of habitats, especially in temperate agricultural pastures, coniferous leaf litters, and coastal vegetations [8]. Among the NTFs, A. oligospora has generally been considered to be the most common nematode predator in temperate soils [9]. In soil environments, A. oligospora naturally encounters a broad range of nematodes and behaves as a generalist predator with the characteristic ability of forming adhesive trapping nets when its mycelia are in contact with nematodes. Unlike endoparasitic fungi, NTFs are able to grow saprophytically. However, how they maintain and balance their saprophytic and predatory lifestyles are not known. In addition, how geographic populations interact with each other and how ecological factors impact population dynamics remain poorly understood. Such knowledge is important not only for understanding the diversity and evolution of these fungi but also for identifying the most appropriate isolate(s) for commercial biological-control applications [3][6].
Most ecological studies of nematophagous fungi have been restricted to surveys on their geographical and seasonal distributions, including examining the effects of abiotic (e.g., soil conditions) and biotic (mainly nematode density) factors on their distributions [8][9][10][11][12][13][14][15]. Though molecular techniques have increasingly been used to examine phylogenetic relationships among nematode-trapping Orbiliales [16][17][18][19][20]and to investigate the molecular genetics of fungus–nematode interactions [21][22][23][24][25][26][27][28][29], they have scarcely been used to study the patterns of genetic variation present in NTF populations, including the spatial and temporal distributions of genotypes in specific species. The authors of an earlier study[30] analyzed 97 A. oligospora strains from several geographic locations and ecological niches in China using DNA sequences at three gene fragments: its (internal transcribed spacer region of the ribosomal RNA), tub (β-tubulin), and rpb2 (RNA polymerase II subunit). That study identified a large number of unique alleles and genotypes, as well as their limited geographic distributions, consistent with a large effective population size of A. oligospora and its potential for genetic differentiation among geographic populations, likely driven by ecological adaptation. Further analyses of more samples using restriction fragment length polymorphism (RFLP) genotyping revealed a similar pattern . Recently, the heritability of A. oligospora phenotypic variation in the response to nematodes was assessed by analyzing the genetic variation of genomic SNPs using 18 wild isolates. However, due to limited sampling at most geographic locations and/or the small number of molecular loci used, A. oligospora’s overall patterns of genetic variations, the relationships between genetic variations and phenotypic variations, and its mode of reproduction remain to be fully described.
2. Results
2.1. Genetic Diversity of the Chinese Samples of A. Oligospora Detected by STRs and MLST
Based on 200 initially designed STR primer pairs from the genome of A. oligospora (ATCC 24927), we selected 20 to genotype strains of A. oligospora. A previous study classified the polymorphism level of STR markers based on PIC values into low (PIC < 0.25), medium (0.5 > PIC > 0.25), and high (PIC > 0.5) [51]. Based on this grouping, the 20 STR markers we developed for A. oligospora were found to have a moderate to high level of polymorphism, with 13 having high polymorphism, six having medium polymorphism, and only one having a low PIC value. The highest discriminatory power value for a single locus was obtained with A191, with a gene diversity value of 0.87 and 21 different alleles in our sample.
Among the 239 A. oligospora isolates from 19 different geographical populations, a total of 188 alleles and 178 multilocus genotypes were found based on the 20 STR loci. The number of alleles range from 3 to 21, with an average of 9.4 alleles per locus. Of the 188 alleles, 140 were shared between at least two of the 19 geographical populations in China, the remain 48 alleles were found only in one geographical population each. The 19 geographical populations also differed in their total numbers of alleles, which ranged from 21 (Kanas Lake, Xinjiang) to 88 (Heijing, Yunnan). Except for their absence in three geographic populations (Inner Mongolia, Qinghai, and Kanas Lake, and Xinjiang populations), private alleles were found in all other 16 populations. Of the total 178 STR multilocus genotypes, only four were shared by two or more geographical populations, and the other 174 were only found in one geographical population each. The most shared genotype was found among two populations from Xinjiang and three populations from Dianchi, Yunnan, Sichuan, and Hubei.
2.2. Genetic Differentiation and Population Structure
Though significant genetic differentiations were found among many geographic populations, our analyses also showed evidence of gene flow among certain geographic populations. The existence of gene flow and genetic differentiation was further supported by results from the STRUCTURE analysis that showed that there were two genetic clusters (K = 2) in the Chinese population of A. oligospora, and most geographic populations contained alleles and genotypes of both genetic clusters. The population structure indicated by STRUCTURE software and PCoA was very similar, and both the MLST and STR datasets identified two distinct genetic clusters in all 239 A. oligospora isolates. Specifically, most samples from Southwestern China (Sichuan, Guizhou, Tibet, and Yunnan including Dianchi, Heijing, Yimen, and Gejiu) and Hainan Island formed one cluster, while those from other parts of China formed another cluster. Populations in the former clade were found to have more frequent genetic exchanges than those in the second cluster. However, the composition of two different genetic clusters indicated by the two kinds of molecular markers were quite different in some geographic populations. For example, STRs detected the existence of genetic elements of the second cluster in geographic populations from Hubei, Inner Mongolia, Jilin, Xinjiang, and Guangxi, while they were absent in the MLST dataset. Interestingly, most genotypes containing elements of both genetic clusters were distributed in Southwestern China and Hainan. These genotypes likely represent hybrids of those two genetic clusters.
2.3. Phylogenetic Divergence
Phylogenetic analyses based on SNPs and MLST identified two distinct clades (Ⅰ and Ⅱ) within A. oligospora from 19 geographic populations in China, consistent with the two genetic clusters revealed by the population structure analyses. Clades Ⅰ and Ⅱ of the MLST phylogeny included 38 and 21 multilocus sequence types, respectively. Most A. oligospora samples from different geographic populations (except for Guizhou) were widely distributed across Clade ⅠI. Interestingly, different geographical populations had different distribution patterns between the two clades: five geographic populations (Hubei, Jilin, Guangxi, and two populations from Xinjiang) were only found in Clade ⅠI, and samples from Tibet were only found in Clade I. Two populations (Hainan and Heijing from Yunnan) were widely distributed in both clades.
Significant differences in the relationships among strains among the analyzed gene fragments were identified based on the pairwise comparisons of topologies among all single gene phylogenetic trees (Figures 1). The results indicated evidence of potential allele-sharing and/or hybridization between the genetic lineages of this important NTF. For example, strains containing both blue and red alleles in the STRUCTURE analysis (colored in green on trees) formed tight and independent clusters on trees of mapk, rpb2 and sp fragments, suggesting they belonged to populations that are diverging from the other two clades. However, the same strains had mixed relationships with others based on sequences at its, tub, and tef-1 loci. Together, these differences in phylogenetic relationships among strains by different gene fragments are consistent with recombination in nature.
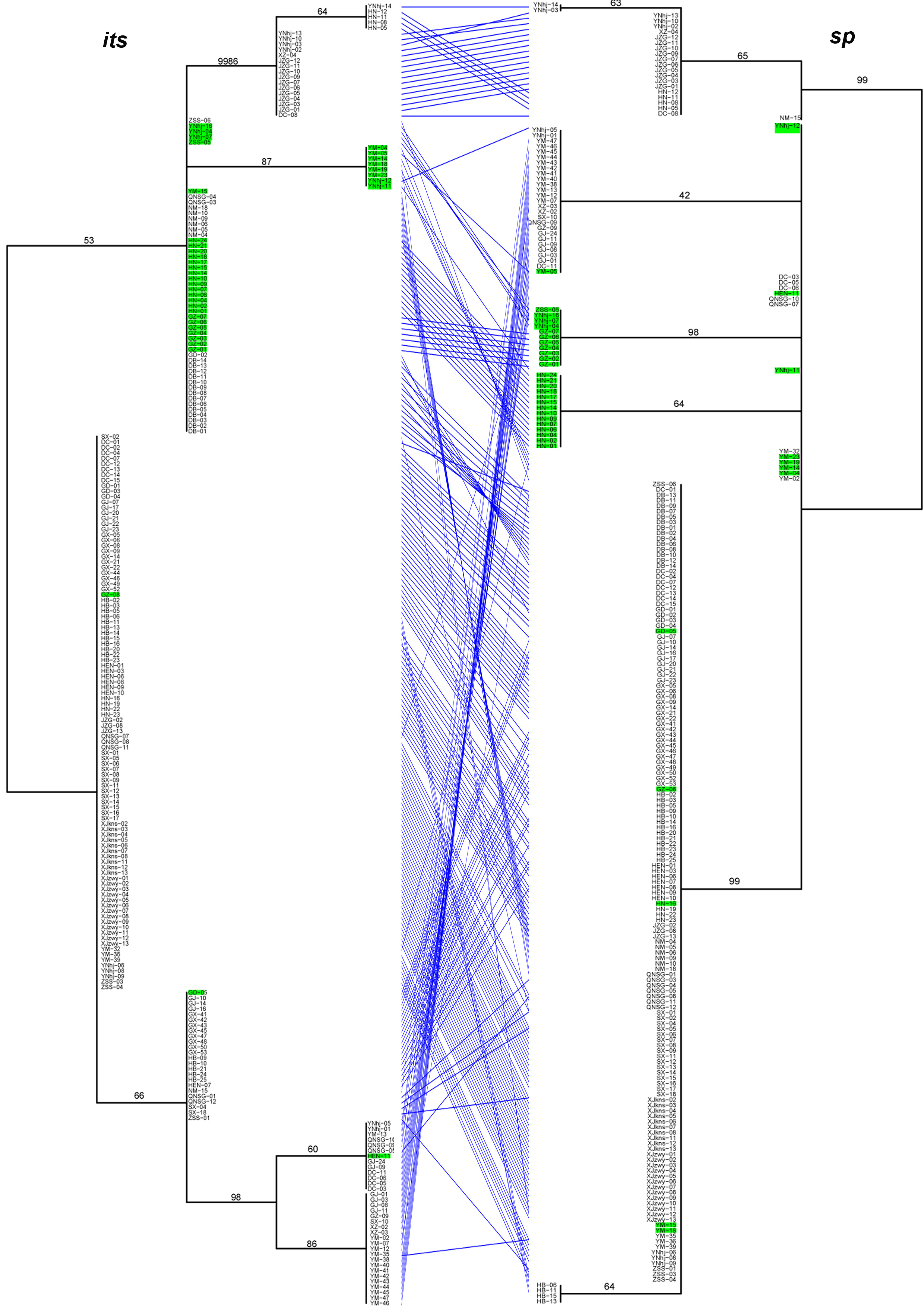
Figure 1. The tanglegram between its and sp phylogenies. Notes: Branch support values are indicated by numbers near branches, and putative hybrids (as suggested by STRUCTURE analysis) are colored in green in the tanglegram.
2.4. Clonality and Recombination
The rBarD and phylogenetic incompatibility tests for detecting recombination were conducted for (i) the total sample, (ii) samples from each geographical sample groups by both the STR and MLST datasets, and (iii) samples representing MLST genotypes in each of the phylogenetic clades (Table 1); the allelic sequences at each of the six MLST loci were treated as alleles in these tests. Though predominantly clonal population structures were detected for these sample sets, we found unambiguous evidences for recombination among the 20 STR loci (PrC = 0.01, p < 0.001; rBarD = 0.355, p < 0.001) and six MLST loci (PrC = 0, p = 1; rBarD = 0.6398, p < 0.001) in the total sample, including all 239 A. oligospora isolates. As indicated by the phylogenetic incompatibility test, variable levels of recombination were found within different sample groups. Interestingly, population groups from Southwestern China had the highest levels of phylogenetic incompatibility. It is worth noting that phylogenetic incompatibility was also identified in all samples where random recombination was rejected, indicating that low levels of recombination are common in samples of A. oligospora in China.
Table 1. Results of multilocus linkage disequilibrium analyses for different sample groups of A. oligospora from China.
|
Sample Groups |
Sample Size |
Phylogenetic Compatibility (p Value) |
rBarD (p Value) |
||
|
MLST |
STR |
MLST |
STR |
||
|
Total |
239 |
0 (1) |
0.01 (<0.001) |
0.6398 (<0.001) |
0.355 (<0.001) |
|
Central |
25 |
0.8667 (0.447) |
0.7 (0.038) |
0.1077 (0.009) |
0.1331 (<0.001) |
|
East |
5 |
1 (1) |
0.9947 (0.096) |
0.6805 (0.001) |
0.3039 (<0.001) |
|
North |
24 |
0.9333 (1) |
0.8 (<0.001) |
0.2784 (<0.001) |
0.2772 (<0.001) |
|
Northeast |
14 |
1 (1) |
0.9211 (0.009) |
-nan * (<0.001) |
0.115 (<0.001) |
|
Northwest |
33 |
0.8 (0.889) |
0.8632 (<0.001) |
0.576 (<0.001) |
0.3988 (<0.001) |
|
South |
46 |
0.4667 (<0.001) |
0.2526 (<0.001) |
0.6867 (<0.001) |
0.288 (<0.001) |
|
Southwest |
92 |
0.2667 (<0.001) |
0.0579 (<0.001) |
0.661 (<0.001) |
0.3874 (<0.001) |
|
Clade I |
151 |
0.1333 (<0.001) |
0.021 (<0.001) |
0.5042 (<0.001) |
0.2019 (<0.001) |
|
Clade II |
46 |
0.5333 (<0.001) |
0.3052 (<0.001) |
0.6379 (<0.001) |
0.4439 (<0.001) |
Notes: Geographic populations included in each of the major regions in China: Central: Hubei, Henan; East: Zhejiang, Shandong; North: Hebei, Nei Mongol; Northeast: Jilin; Northwest: Shaanxi, Qinghai, Xinjiang; South: Guangdong, Guangxi, Hainan; Southwest: Sichuan, Guizhou, Yunnan, Tibet. * All strains in this population have the same haplotypes in six gene fragments. MLST: multilocus sequence typing; STR: short tandem repeat; rBarD: standardized index of association.
2.5. Phenotypical Characterization
2.5.1. Conidial Morphology
For conidial morphology analyses, 38 strains of A. oligospora—including 30, 6, and 2 strains from the divergent Clades A, B, and C identified in our previous study [30], respectively—were selected to observe. Clades A and B respectively correspond to our Clades Ⅰ and Ⅱ here, and Clade C falls into the basal branch of Clade Ⅱ. After conidia sporulation on CMA, the length and width of 50 mature conidia of each strain were observed and measured, and the length–width (L/W) ratio was selected as the quantitative measure for the following statistical analyses.
First, we tested variance homogeneity among the data from three different phylogenetic clusters. A Levene’s p value of greater than 0.05 was obtained, suggesting there was no significant difference in the total variance among the three clusters. Second, an analysis of variance was conducted. The L/W ratio of Clade C strains was found to be significantly higher than those of Clades A and B by Duncan’s multiple comparison test (Figure 2c). The L/W ratios of strains from Clades A and B were not significantly different from each other. Together, our results indicated some morphological differentiation among the clades within A. oligospora strains from China.
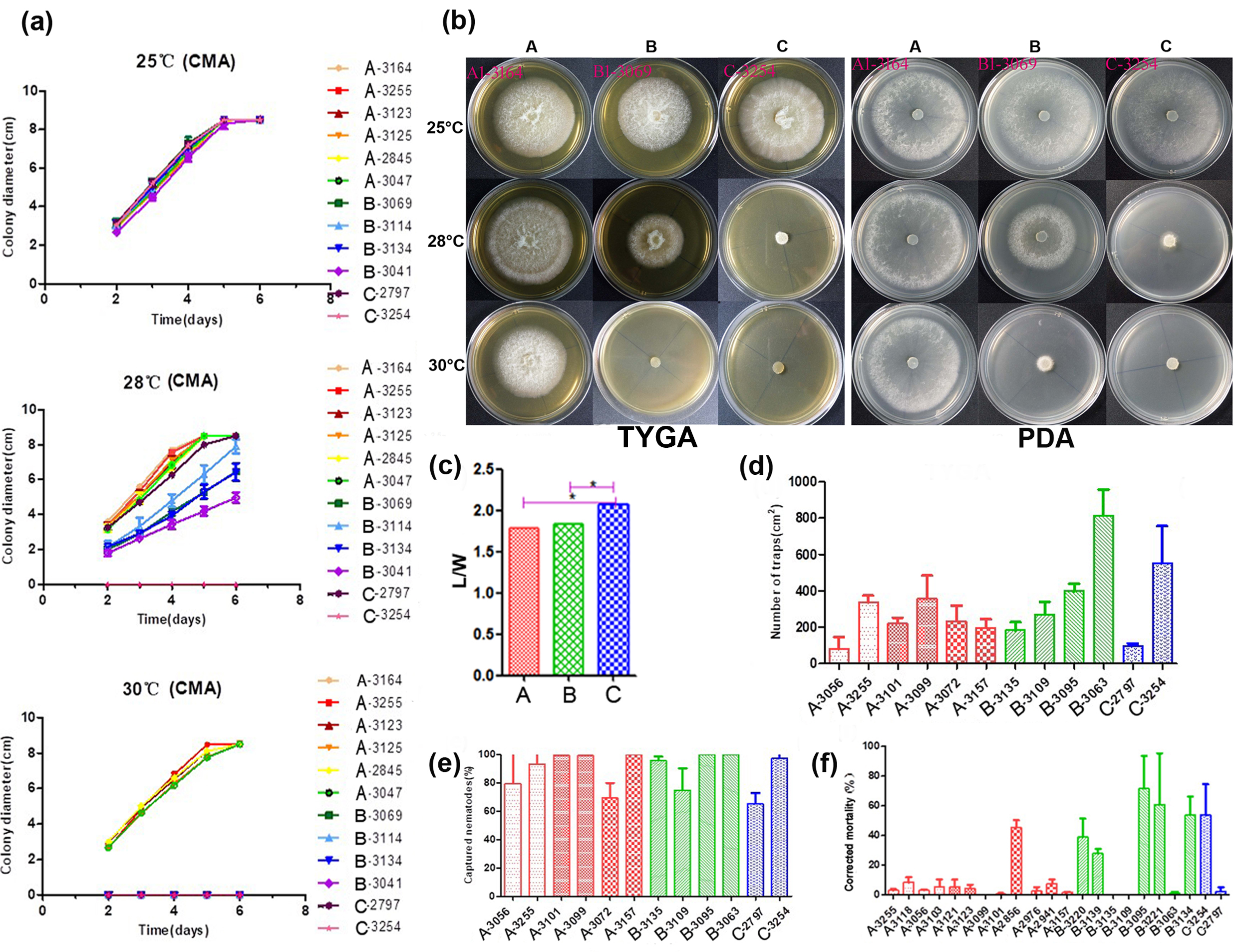
Figure 2. Phenotypical characterization of strains from the three main lineages. (a) The growth state of 12 representative A. oligospora strains incubation at 25, 28, and 30 °C on CMA. (b) Colony morphology of representative strains at 25, 28, and 30 °C on TYGA and PDA plates. (c) Pairwise comparisons of L/M among representative strains. (d) Comparison of trap formation, (e) captured nematodes, and (f) nematicidal activities of fermentation broth among representative strains. Notes: CMA: corn meal agar, TYGA: the tryptone, yeast extract, and glucose agar, PDA: potato dextrose agar, * significantly different.
2.5.2. Mycelial Growth
A total of 12 strains representing three clades (six from Clade A, four from Clade B, and two from Clade C) from different geographic populations were selected to investigate their potential differences in mycelial growth rate and colony morphology on three different media at 25, 28, and 30 °C. The fungal colonies on the CMA medium were generally very loose and their aerial hyphae were sparse. By contrast, those on TYGA medium produced extremely dense mycelia and their aerial mycelia grew most robustly. The growth status of hyphae on the PDA medium was intermediate between CMA and TYGA (Figure 2b).
Overall, the optimal growth temperature of different A. oligospora strains was 25 °C. At 25 °C, all strains could grow normally and showed the fastest growth rate among the three tested temperatures. However, the growth rates of isolates from Clades B and C (except YMF1.02797 from Clade C) were slower than those of Clade A at 28 °C, and the aerial mycelia deviated from radial spread. In fact, isolates from Clades B and C were unable to grow at 30 °C (Figure 2a,b). Together, these results suggest clade-specific differences in colony morphology and mycelial growth, regardless of geographic origins.
2.5.3. Conidial Yield, Trap Formation and Nematode-Trapping Bioassay
A. oligospora, which can capture nematodes by forming three-dimensional networks from specialized hyphae, is considered the most abundant nematode predator in temperate soils [11]. In addition, genetic differences have been reported to have effects on the response of A. oligospora to its prey, though robust correlation has not been observed [21]. Therefore, the conidial yield, trap formation, and nematode-trapping bioassays were further investigated to examine whether there were notable differences among nematicidal activities among the three phylogenetic clades identified in a previous study.
Overall, we found significant differences in conidial yield among strains, but the differences among phylogenetic clades were not statistically significant. Strains from each of the phylogenetic clades have variable abilities to produce conidia. For example, three strains from the three clades (with one representing each clade)—YMF1.03101 (Clade A), YMF1.03135 (Clade B) and YMF1.03254 (Clade C)—were found to have significantly higher conidial yields than all other tested strains from either the same or different clades.
The number of traps produced by different strains of A. oligospora was most distinguishing when exposed to C. elegans for 12 h. The highest number was produced by strain YMF1.03063 of Clade B at >47% more than others, while the numbers produced by YMF1.03056 of Clade A and YMF1.02797 of Clade C were the least, only accounting for about 20% of that produced by YMF1.03063. The number of trappers produced by other tested strains were similar (Figure 2d). Overall, there was no significant correlation between the various abilities of the trap formation and phylogenetic division of A. oligospora strains from China. Correspondingly, limited difference regarding nematode-trapping abilities was found among the three phylogenetic clusters (Figure 2e).
We further detected the pathogenicity of fermentation broth from different representative strains in the three phylogenetic clades. Forty-eight hours after the nematodes were added into the fermentation supernatants, the lethal effects of different A. oligospora strains on nematodes was significantly different. Generally, strains YMF1.02856 (Clade A), YMF1.03095 (Clade B), and YMF1.03254 (Clade C) had obvious nematicidal effect, and the corrected mortality caused by strain YMF1.03095 was as high as 71.5%. However, strains YMF1.03056 and YMF1.02797, which showed the distinguishing ability to form traps and capture nematodes on the solid media, produced fermentation supernatants with weak pathogenicity (Figure 2f). Overall, we found no significant difference among clades in terms of the nematicidal activity of their fermentation broth.
3. Discussion
3.1. Development of Novel STRs for NTF
For decades, molecular tools have acted as complementary techniques to differentiate species and examine phylogenetic relationships and systematics for NTF with traditional morphological methods[17][18][19][20][22]. Recently, MLST and RFLP have been used for ecological studies of nematophagous fungi , and Orbiliales-specific PCR primers were developed to directly detect NTF in environmental samples. However, limited information is available on the patterns of genetic variations within and among geographic populations. This study described a new panel of 20 STRs for the exact and high-resolution genotyping strains of A. oligospora from China. About 2/3 of these STR markers were identified as highly polymorphic, and abundant allelic and genotypic diversities were detected among isolates of A. oligospora from China, thus indicating the high discriminatory power of our newly developed STRs.
Traditionally, the sprinkle plates method is used to isolate and identify NTFs based on their colony morphology and type of nematode traps produced. However, it is often difficult to analyze such data to infer the relationships among strains and populations. For example, the sprinkle plate method may be biased toward detecting species of nematophagous fungi that are adapted for growth in culture and have good predacious ability [31]. The STR markers were highly discriminatory for identifying alleles and genotypes, and they enabled the generation of reproducible research results that could be easily shared among investigators when analyzing NTFs. Furthermore, the interactions among fungi, nematodes, and other soil organisms in different soils and the population dynamics of NTFs could be better uncovered with these markers.
In this study, the overall genetic clustering and phylogenetic separation indicated by the two kinds of molecular marks (STR vs. MLST) were similar. However, differences concerning genetic differentiation and the level of genetic exchange (hybridization) were also found. Specifically, more genetic variation among populations and frequent gene exchanges were revealed by STRs, which was likely due to the faster evolutionary rate of STRs than MLST. Indeed, the STR markers allowed for the detection of hybridization in most geographic populations, suggesting the hybridization events likely occurred very recently. Similarly, STRs failed to identify a statistically significant positive correlation between genetic and geographic distances or to discriminate the ancient population divergence that the MLST revealed. In addition, though the STR markers showed overall higher population differentiation than the MLST markers, they also showed greater gene flow among many of the 19 geographical populations. Together, due to the high mutation rate, the STR markers allowed us to capture the recent, fine-scale population genetic events that the MLST markers failed to reveal. Future studies should focus on analyzing the potential factors contributing to the differences in population structures, as revealed by MLST and STR markers, including factors such as ecological niche adaptation and host nematode distributions and preferences.
3.2. Historical Population Differentiation
This study identified two distinctly differentiated genetic clusters among A. oligospora strains in China, indicating historical population divergence in this species. One of the major genetic cluster was composed of samples from Southwestern China and Hainan Island. Geographic populations from this region had a large number of private genotypes and unique alleles. The widespread genetic diversity and uniqueness in these areas suggested historical geographic isolation and, likely, the accumulation of locally adapted genotypes within and among local populations. Factors such as landscape features (e.g., mountain ranges), climate, and habitats in different geographic populations could all impact local ecology, genetic drift, selection, and adaptation. Specifically, Southwest China has a large number of mountain ranges and river systems that create a diversity of ecological niches, making it one of the most important biodiversity hot spots on earth [32]. Indeed, the high species diversity and endemism of fungal species and genotypes have been frequently revealed in this area [33]. Furthermore, Hainan is the only island population with a tropical monsoon oceanic climate, and the geographic isolation between Hainan and the rest of China may result in the accumulation of unique genetic diversity. Together, the geographic and climatic factors likely contribute to the observed unique genetic diversities within individual local populations.
3.3. Recent Hybridization and Recombination
Study analyses revealed evidence for genetic exchanges among the strains and populations in Southwestern China and Hainan Island. As indicated by the population structure and gene genealogical analyses, there is extensive evidence for allele-sharing among lineages of A. oligospora, most likely due to hybridization. In addition to inter-lineage hybridization, unambiguous evidence for recombination within individual lineages, though most of the samples showed evidence of non-random recombination. The frequent sharing of alleles but limited or no overlap in multilocus genotypes were further consistent with frequent recombination in nature.
Indeed, the widespread distributions of both mat1-1 and mat1-2 idiomorphs across geographic populations made mating between strains with different genetic elements possible. The finding that a couple of strains with different mating types shared the same STR genotype is also a strong piece of evidence for their recombinant origins. Results here complement those in an earlier study that showed A. oligospora populations from stressful environments to be in greater linkage equilibrium than those from other environments in the same geographic areas .
3.4. Intraspecies Diversification
The Fungal kingdom is among the most diverse and specious groups of eukaryotes. At present, we are still far from knowing the full extent of fungal diversity. One major finding based on DNA sequences over the last two decades has been the existence of multiple genetically divergent cryptic species within many previously defined species [39][40][41]. Within NTF, morphology-based classifications of strains and species have also likely underestimated the true magnitude of species diversity in this group of fungi. Though the globally isolated 22 isolates of a promising biocontrol agent Duddingtonia flagrans have a single recent common ancestor with limited genetic differentiation among geographic populations , several species within the NTF genera Arthrobotrys and Monacrosporium have been found to contain significant genetic variation and cryptic species among morphologically similar isolates.
Over the last two decades, the phylogenetic species concept (PSC) has been frequently used for identifying fungal species . According to the PSC, species are diagnosed as a cluster of individuals that are sufficiently differentiated from other clusters, as revealed by DNA sequences. Given the presence of genetically differentiated clusters of strains in the phylogenetic tree, we sought to further determine whether the observed genetic divergence within A. oligospora was associated with phenotypic characters such as morphology and nematode-trapping abilities. As indicated by shape of conidia (L/W), the morphological differentiation of A. oligospora strains was found to be associated with phylogenetic lineage differentiations. However, large variations among strains are also observed within each of the lineages, a result consistent with significant observed genetic variations within individual lineages of this species. The observed variations suggest in field applications as biocontrol agents against plant-parasitic nematodes, a mixture of genotypes from the locally sourced strains might be more effective than a single strain.
The genetic clusters and phylogenetic lineages identified here represent speciating populations within A. oligospora. At the same time, the identification of hybrids in most geographic populations suggests that strains in different clusters/lineages are sexually compatible. Indeed, the comparable distributions of mat1-1 and mat1-2 genes suggest that sexual mating is likely common in natural populations of this species. Analyses of laboratory crosses between strains from the same and different lineages could help reveal the underlying genetic mechanisms responsible for the observed phenotypic variations. Host preference has been found in pairs of tightly interacting hosts and pathogens due to shared common evolutionary histories. However, in facultative pathogens, the selective pressure exerted by both hosts and habitats could be equally important [42][43]. At present, the extent of nematode-host specificity and how such specificity may be related to phenotypic diversification and ecological niche adaptation remain largely unknown.
References
- Abad, P.; Gouzy, J.; Aury, J.-M.; Castagnone-Sereno, P.; Danchin, E.G.J.; Deleury, E.; Perfus-Barbeoch, L.; Anthouard, V.; Artiguenave, F.; Blok, V.C., et al. Genome sequence of the metazoan plant-parasitic nematode Meloidogyne incognita. Nature Biotech. 2008, 26, 909-915, doi:10.1038/nbt.1482.
- Giannakou, I.O.; Anastasiadis, I.A.; Gowen, S.R.; Prophetou-Athanasiadou, D. Effects of a non-chemical nematicide combined with soil solarization for the control of root-knot nematodes. Crop Prot. 2007, 26, 1644-1654, doi:10.1016/j.cropro.2007.02.003.
- Zhang, Y.; Li, S.; Li, H.; Wang, R.; Zhang, K.Q.; Xu, J. Fungi-Nematode Interactions: Diversity, Ecology, and Biocontrol Prospects in Agriculture. J. Fungi (Basel) 2020, 6, doi:10.3390/jof6040206.
- Jiang, X.; Xiang, M.; Liu, X. Nematode-Trapping Fungi. Microbiol Spectr 2017, 5, doi:10.1128/microbiolspec.FUNK-0022-2016.
- Yang, J.; Wang, L.; Ji, X.; Feng, Y.; Li, X.; Zou, C.-G.; Xu, J.; Ren, Y.; Mi, Q.; Wu, J., et al. Genomic and Proteomic Analyses of the Fungus Arthrobotrys oligospora Provide Insights into Nematode-Trap Formation. PLoS pathogens 2011, 7, e1002179, doi:10.1371/journal.ppat.1002179.
- Zhang, Y.; Zhang, K.-Q.; Hyde, K. The Ecology of Nematophagous Fungi in Natural Environments. In Nematode-Trapping Fungi, Zhang, K.-Q., Hyde, K.D., Eds. Springer Netherlands: Dordrecht, 2014; 10.1007/978-94-017-8730-7_4pp. 211-229.
- Li, J.; Zou, C.; Xu, J.; Ji, X.; Niu, X.; Yang, J.; Huang, X.; Zhang, K.Q. Molecular mechanisms of nematode-nematophagous microbe interactions: basis for biological control of plant-parasitic nematodes. Annu. Rev. Phytopathol. 2015, 53, 67-95, doi:10.1146/annurev-phyto-080614-120336.
- Gray, N.F. Ecology of nematophagous fungi: distribution and habitat. Ann. Appl. Biol. 1983, 102, 501-509, doi:10.1111/j.1744-7348.1983.tb02721.x.
- Niu, X.-M.; Zhang, K.-Q. Arthrobotrys oligospora: a model organism for understanding the interaction between fungi and nematodes. Mycology 2011, 2, 59-78, doi:10.1080/21501203.2011.562559.
- Waghorn, T.S.; Leathwick, D.M.; Chen, L.Y.; Gray, R.A.; Skipp, R.A. Influence of nematophagous fungi, earthworms and dung burial on development of the free-living stages of Ostertagia (Teladorsagia) circumcincta in New Zealand. Vet. Parasitol. 2002, 104, 119-129, doi:10.1016/s0304-4017(01)00629-x.
- Gray, N. Nematophagous fungi with particular reference to their ecology. Biol. Rev. Cambri. Philosophi. Soc. 1987, 62, 245-304, doi:10.1111/j.1469-185X.1987.tb00665.x.
- Pathak, E.; Campos–Herrera, R.; El–Borai, F.E.; Duncan, L.W. Spatial relationships between entomopathogenic nematodes and nematophagous fungi in Florida citrus orchards. J. Inverteb. Pathol. 2017, 144, 37-46, doi:10.1016/j.jip.2017.01.005.
- Jaffee, B.A.; Strong, D.R.; Muldoon, A.E. Nematode-trapping fungi of a natural shrubland: Tests for food chain involvement. Mycologia 2018, 88, 554-564, doi:10.1080/00275514.1996.12026686.
- Deng, W.; Wang, J.L.; Scott, M.B.; Fang, Y.H.; Liu, S.R.; Yang, X.Y.; Xiao, W. Sampling methods affect Nematode-Trapping Fungi biodiversity patterns across an elevational gradient. BMC Microbiol. 2020, 20, 15, doi:10.1186/s12866-020-1696-z.
- Strom, N.; Hu, W.; Haarith, D.; Chen, S.; Bushley, K. Interactions between soil properties, fungal communities, the soybean cyst nematode, and crop yield under continuous corn and soybean monoculture. Appl. Soil Ecol. 2020, 147, 103388, doi:10.1016/j.apsoil.2019.103388.
- Li, Y.; Hyde, K.D.; Jeewon, R.; Cai, L.; Vijaykrishna, D.; Zhang, K. Phylogenetics and evolution of nematode-trapping fungi (Orbiliales) estimated from nuclear and protein coding genes. Mycologia 2005, 97, 1034-1046.
- Rubner, A. Revision of predacious hyphomycetes in the Dactylella-Monacrosporium complex. Stud. Mycol. 1996, 39, 1-134.
- Scholler, M.; Hagedorn, G.; Rubner, A. A reevaluation of predatory orbiliaceous fungi. II. A new generic concept. Sydowia 1999, 51, 89-113.
- Yang, E.; Xu, L.; Yang, Y.; Zhang, X.; Xiang, M.; Wang, C.; An, Z.; Liu, X. Origin and evolution of carnivorism in the Ascomycota (fungi). Proc. Natl. Acad. Sci. USA 2012, 109, 10960-10965, doi:10.1073/pnas.1120915109.
- Yang, Y.; Yang, E.C.; An, Z.Q.; Liu, X.Z. Evolution of nematode-trapping cells of predatory fungi of the Orbiliaceae based on evidence from rRNA-encoding DNA and multiprotein sequences. Proc. Natl. Acad. Sci. USA 2007, 104, 8379-8384.
- Yang, C.T.; Vidal-Diez de Ulzurrun, G.; Goncalves, A.P.; Lin, H.C.; Chang, C.W.; Huang, T.Y.; Chen, S.A.; Lai, C.K.; Tsai, I.J.; Schroeder, F.C., et al. Natural diversity in the predatory behavior facilitates the establishment of a robust model strain for nematode-trapping fungi. Proc. Natl. Acad. Sci. USA 2020, 117, 6762-6770, doi:10.1073/pnas.1919726117.
- Liu, M.; Cheng, X.; Wang, J.; Tian, D.; Tang, K.; Xu, T.; Zhang, M.; Wang, Y.; Wang, M. Structural insights into the fungi-nematodes interaction mediated by fucose-specific lectin AofleA from Arthrobotrys oligospora. Int. J. Bio. Macromol. 2020, 164, doi:10.1016/j.ijbiomac.2020.07.173.
- Kuo, T.H.; Yang, C.T.; Chang, H.Y.; Hsueh, Y.P.; Hsu, C.C. Nematode-Trapping Fungi Produce Diverse Metabolites during Predator-Prey Interaction. Metabolites 2020, 10, doi:10.3390/metabo10030117.
- Ji, X.; Yu, Z.; Yang, J.; Xu, J.; Zhang, Y.; Liu, S.; Zou, C.; Li, J.; Liang, L.; Zhang, K.Q. Expansion of Adhesion Genes Drives Pathogenic Adaptation of Nematode-Trapping Fungi. iScience 2020, 23, 101057, doi:10.1016/j.isci.2020.101057.
- Basso, M.F.; Lourenco-Tessutti, I.T.; Mendes, R.A.G.; Pinto, C.E.M.; Bournaud, C.; Gillet, F.X.; Togawa, R.C.; de Macedo, L.L.P.; de Almeida Engler, J.; Grossi-de-Sa, M.F. MiDaf16-like and MiSkn1-like gene families are reliable targets to develop biotechnological tools for the control and management of Meloidogyne incognita. Sci. Rep. 2020, 10, 6991, doi:10.1038/s41598-020-63968-8.
- Liang, L.M.; Zou, C.G.; Xu, J.; Zhang, K.Q. Signal pathways involved in microbe-nematode interactions provide new insights into the biocontrol of plant-parasitic nematodes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2019, 374, 20180317, doi:10.1098/rstb.2018.0317.
- Wang, R.; Dong, L.; He, R.; Wang, Q.; Chen, Y.; Liangjian, Q.; Zhang, Y.-A. Comparative genomic analyses reveal the features for adaptation to nematodes in fungi. DNA Research 2018, 25, doi:10.1093/dnares/dsx053.
- Hsueh, Y.P.; Gronquist, M.R.; Schwarz, E.M.; Nath, R.D.; Lee, C.H.; Gharib, S.; Schroeder, F.C.; Sternberg, P.W. Nematophagous fungus Arthrobotrys oligospora mimics olfactory cues of sex and food to lure its nematode prey. eLife 2017, 6, e20023 doi:10.7554/eLife.20023.
- Wang, X.; Li, G.H.; Zou, C.G.; Ji, X.L.; Liu, T.; Zhao, P.J.; Liang, L.M.; Xu, J.P.; An, Z.Q.; Zheng, X., et al. Bacteria can mobilize nematode-trapping fungi to kill nematodes. Nature Commun. 2014, 5, 5776, doi:10.1038/ncomms6776.
- Zhang, Y.; Yu, Z.-F.; Xu, J.; Zhang, K.-Q. Divergence and dispersal of the nematode-trapping fungus Arthrobotrys oligospora from China. Environ. Microbio. Rep. 2011, 3, 763-773, doi:10.1111/j.1758-2229.2011.00297.x.
- Bailey, F.; Gray, N.F. The comparison of isolation techniques for nematophagous fungi from soil. Ann. Appl. Bio. 1989, 114, 125-132.
- Myers, N.; Mittermeier, R.A.; Mittermeier, C.G.; da Fonseca, G.A.; Kent, J. Biodiversity hotspots for conservation priorities. Nature 2000, 403, 853-858, doi:10.1038/35002501.
- Feng, B.; Yang, Z. Studies on diversity of higher fungi in Yunnan, southwestern China: A review. Plant Divers. 2018, 40, 165-171, doi: 10.1016/j.pld.2018.07.001.
- Zhou, D.; Wang, R.; Li, X.; Peng, B.; Yang, G.; Zhang, K.-Q.; Zhang, Y.; Xu, J. Genetic Diversity and Azole Resistance Among Natural Aspergillus fumigatus Populations in Yunnan, China. Microb. Ecol. 2021, 10. doi:10.1007/s00248-021-01804-w.
- Zhou, D.; Korfanty, G.A.; Mo, M.; Wang, R.; Li, X.; Li, H.; Li, S.; Wu, J.Y.; Zhang, K.Q.; Zhang, Y., et al. Extensive Genetic Diversity and Widespread Azole Resistance in Greenhouse Populations of Aspergillus fumigatus in Yunnan, China. mSphere 2021, 6, e00066-21, doi:10.1128/mSphere.00066-21.
- Wu, J.-Y.; Guo, H.; Wang, H.-M.; Yi, G.-H.; Zhou, L.-M.; He, X.-W.; Zhang, Y.; Xu, J. Multilocus sequence analyses reveal extensive diversity and multiple origins of fluconazole resistance in Candida tropicalis from tropical China. Sci. Rep. 2017, 7, 42537, doi:10.1038/srep42537.
- Fang D.Z.; Liu, X.L.; Chen X.R.; Yan, W.R.; He, Y.L.; Cheng, Y.; Chen, J.; Li, Z.M.; Guo, L.T.; Wang, T.H., et al. Fusarium Species and Fusarium oxysporum Species Complex Genotypes Associated With Yam Wilt in South-Central China. Front. Microbiol. 2020, 11, 1964, doi:10.3389/fmicb.2020.01964.
- Cao, Y.; Zhang, Y.; Yu, Z.; Mi, F.; Liu, C.; Tang, X.; Long, Y.; He, X.; Wang, P.; Xu, J. Structure, Gene Flow, and Recombination among Geographic Populations of a Russula virescens Ally from Southwestern China. PLoS One 2013, 8, e73174, doi:10.1371/journal.pone.0073174.
- Xu, J. Fungal Species Concepts in the Genomics Era. Genome 2020, 63, doi:10.1139/gen-2020-0022.
- Guo, T.; Wang, H.C.; Xue, W.Q.; Zhao, J.; Yang, Z.L. Phylogenetic Analyses of Armillaria Reveal at Least 15 Phylogenetic Lineages in China, Seven of Which Are Associated with Cultivated Gastrodia elata. PLoS One 2016, 11, e0154794, doi:10.1371/journal.pone.0154794.
- Peintner, U.; Kuhnert-Finkernagel, R.; Wille, V.; Biasioli, F.; Shiryaev, A.; Perini, C. How to resolve cryptic species of polypores: an example in Fomes. IMA fungus 2019, 10, 17, doi:10.1186/s43008-019-0016-4.
- St Leger, R.J.; Screen, S.E.; Shams-Pirzadeh, B. Lack of host specialization in Aspergillus flavus. Appl. Environ. Microbiol. 2000, 66, 320-324, doi:10.1128/aem.66.1.320-324.2000.
- Bidochka, M.J.; Kamp, A.M.; Lavender, T.M.; Dekoning, J.; De Croos, J.N.A. Habitat association in two genetic groups of the insect-pathogenic fungus Metarhizium anisopliae: uncovering cryptic species? Appl. Env. Microbiol. 2001, 67, 1335-1342.