Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Conner Chen and Version 1 by Diego Laderach.
Current data indicates that anti-tumor T cell-mediated immunity correlates with a better prognosis in cancer patients. However, it has widely been demonstrated that tumor cells negatively manage immune attack by activating several immune-suppressive mechanisms.
- galectins
- tumor immune evasion
- cancer immunotherapy
1. Introduction
In humans, the immune system is constituted by approximately 1013 T lymphocytes at a given time [1]. However, lymphoid homeostasis is highly dynamic, depending on the continuous production of naïve cells in central hematopoietic organs, their activation, survival, and generation of immune memory in peripheral organs. Each of these processes depends on continuous biological signals that lymphocytes receive from their microenvironment [2]. At a molecular level, multiple signaling pathways are involved in lymphocyte regulation. Although we only have a partial understanding of this subject, an extensive compendium of the literature supports galectins as being prominent actors in the regulation of immune homeostasis in both central and peripheral organs (as discussed below).
Currently, 12 human galectin members have been described and found in GenBank (https://www.ncbi.nlm.nih.gov/genbank/; accessed on 15 July 2021). Galectins are involved in the regulation of different cellular processes, among which we can mention cell differentiation, cell adhesion and migration; gene transcription and RNA splicing; and cell cycle and apoptosis [3,4]. Such biological functions depend on their lectinic properties (recognition of N-acetyllactosamine sequences displayed on the cell surface, in the extracellular matrix, or intracellular glycoconjugates) (Table 1). Besides, and studied to a lesser extent, they also depend on non-lectinic properties mediated by interactions of galectins with nucleic acids, proteins, lipids, and complex biomolecules (reviewed in [4], Table 1).
Table 1. The galectins’ interactors and their impact on cancer immune escape.
| Member | Recognition Motif | Interactors | Described Biological Effects | References |
|---|---|---|---|---|
| Galectin-1 | Carbohydrate-dependent Long poly-N-acetyllactosamine chains with a terminal β-galactose residue Carbohydrate-independent |
Proteins of the extracellular matrix: Laminin, fibronectin, collagen, vitronectin, thrombospondin, osteopontin Membrane receptors: Neuropilin-1, VEGF-R2, integrins α1β1 and αMβ2, actin, CD43, CD3,CD4, CD2, CD7, and CD45 Protocadherin-24 GM1 H-RAS Gemin-4, Transcription Factor II-I (TFII-I), snRNP |
Involved in ligand crosslinking and the formation of cell surface lattices Induction and maintenance of multiple signaling pathways Modulation of cell adhesion and migration Modulation of vascular function and immune cell migration Modulation of T cell activation, survival and acquisition of effector functions Regulation Wnt signaling Modulation of Treg function Enhance signaling through GTP-H-RAS nanoscale signaling hubs Nuclear splicing of pre-mRNA and control of gene expression |
[43,44] [45,46,47] [48,49,50] [51,52,53,54] [55,56,57,58,59,60] [61] [62,63] [64,65] [66,67,68,69,70] |
| Galectin-3 | Carbohydrate-dependent Repeating [-3Galβ1-4GlcNAcβ1-]n or poly-N-acetyllactosamine sequences regardless of the presence of a terminal β-galactose residue Carbohydrate-independent |
Proteins of the extracellular matrix: Laminin, vitronectin, collagens I and IV Soluble cytokines Membrane receptors: TCR complex, CD45, CD71, LFA-1, MCAM, TLR-4 LAG-3, VEGF-R2 Nuclear mitotic aparatus (NuMa) K-RAS β-catenin, Protocadherin-24 Endosomal sorting complex required for transport (ESCRT) or Alix Centrin-2 in basal bodies and centrosomas Synexin, CD95 (APO-1/FAS) Cyclin D1/CDK4 complex, hTERT, ATP synthase Gemin-4, snRNP, transcription factor II-I (TFII-I) |
Involved in ligand crosslinking and the formation of cell surface lattices Impact on induction and maintenance of multiple signaling pathways: cell adhesion and migration Reduces cell migration by trapping cytokines Modulation of T cell migration, activation, and functional secretory synapse Cell division Determine membrane nanostructure: impact on signal transduction Wnt signaling TCR downregulation, EGFR trafficking, biogenesis of multivesicular bodies-Exosomes Microtubule organization Apoptosis Cell cycle and senescence Nuclear splicing of pre-mRNA and control of gene expression |
[43,44] [50,71,72] [72] [73,74,75,76,77,78,79,80] [81] [82] [61,83,84] [85,86,87,88,89] [90] [91,92] [93,94,95] [67,68,69,70,96,97] |
| Galectin-7 | Internal or terminal LacNAc repeats | Tid1 Bcl-2 Smad 3 |
Tid1 regulates the nuclear translocation of Galectin-7: role in tumorigenesis and metastasis Sensitize mitochondria to apoptosis signals Decrease expression of TGF-β responsive genes |
[98] [99] [100] |
| Galectin-8 | Human blood groups A and B glycans and sialylated lactose or lactosamine |
Proteins of the extracellular matrix: Laminin, fibronectin, vitronectin, collagen IV Integrins (α3β1 and α6β1), CD166, podoplanin, CD44 NDP52 |
Modulation of cell adhesive and signaling properties Regulation of cell adhesion to endothelium and migration Regulation of apoptosis and inflammation Regulation of bacteria-specific autophagy |
[50,101] [102,103,104,105] [106] [107,108] |
| Galectin-9 | Poly N-acetyllactosamine units | TIM-3, PD-1, CD44, VISTA, 4-1BB, CD40, DR3 Cell surface protein disulfide isomerase, β3 integrin IgE Glut-2 NF-IL6 transcription factor |
Regulation of T-helper 1 cell immunity and tolerance induction Regulation of the redox environment and T cell migration Anti-allergic effects Determines Glut-2 cell-surface half-life, metabolism regulation Regulation of inflammatory cytokines |
[109,110,111,112,113,114,115,116,117] [118] [119] [120] [121] |
Interestingly, the expression profile of galectins is altered during several pathological conditions (expert opinions for each disease have been reviewed in [5,6,7,8,9,10,11,12,13,14]). In particular, the production of galectins-1, -3, -7, -8, and -9 is deregulated during tumorigenesis [15,16,17]. These galectins are detected in transformed cells and their associated stroma in various types of cancers [18,19]. In the latter case, it is not clear whether stromal cells produce galectins by themselves or take them up from neighboring transformed cells [20,21,22]. However, galectins play essential roles in controlling the tumor microenvironment properties independently of their cellular origin [18,19]. Notably, tumor-associated hypoxia and inflammation contribute to the deregulation of galectins expression [23,24,25,26]. As a consequence, increased levels of these galectins are detected in the sera of cancer patients. Until mid-2021, more than 55 independent studies have been recorded in this field (here, and due to space limitations, we only cite some examples on each galectin member [27,28,29,30,31,32,33,34,35,36,37,38]).
Increased galectins production in cancers has been proposed as a prognostic factor since they can easily be detected in the biological fluids and their expression in the tumor microenvironment generally predicts a poor clinical outcome for patients [15,19,39,40,41]. Besides being used as interesting biomarkers, these tumor-derived galectins display active roles in defining tumor progression. Despite the multiple influences these proteins have on the behavior of tumor cells (e.g., tumorigenesis, metastasis, and angiogenesis) [16], it is clear that they constitute an important strategy applied by tumors to evade an immune attack [42]. This review aims to deepen our knowledge on how galectins, abundantly produced by tumors, can impact the production, activation, and effector function of anti-tumor T lymphocytes; some of these processes occur at anatomic sites distant from the tumor (Figure 1).
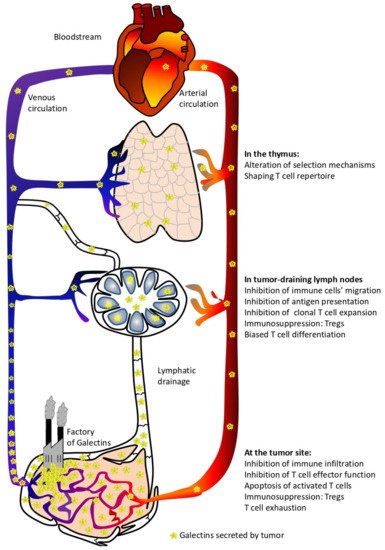
Figure 1. Local and systemic effects of circulating tumor-derived galectins.
1.1. Do Circulating Tumor-Derived Galectins Have Any Impact on Naïve T Cell Production?
From the approximately 2–7 × 107 naïve T cells produced in the thymus every day during adulthood [122,123], very few cells are specific against self-components due to the relative efficiency of the negative selection process [124]. However, if the cancer immunosurveillance theory is correct (which is supported by the low frequency of tumor occurrence throughout our lives) [125], lymphocytes with anti-self-reactivity, available in the periphery in healthy individuals, may be enough to kill cells early after their malignant transformation. However, this optimistic scenario changes radically when transformed cells escape to immune pressure and become less immunogenic. During this tumor development phase, severe impairment of intrathymic T cell differentiation/maturation has been reported, leading to a paralysis of cellular anti-tumor immunity [126,127,128,129,130]. Simultaneously, tumors produce large amounts of galectins (mainly galectins -1, -3, -7, -8, and -9). As already stated, increased levels of galectins in patients’ sera have been reported for several cancer types [27,28,29,30,31,32,33,34,35,36,37,38]. In such conditions of abundant production and secretion, galectins circulate through biological fluids and reach the thymus. Indeed, it was demonstrated that under stress conditions, high levels of circulating galectin-1 are associated with increased numbers of galectin-1-positive cells in the thymus medulla. This effect is not genetically mediated since no upregulation of galectin-1 mRNA was observed in thymic cells [131,132]. In this context, do galectins produced by the multiple cell types within a tumor impact T cell production and education in the thymus?
Since thymic education involves a high rate of cell death [133], galectins’ role in the regulation of thymic apoptosis awakes a particular interest. There exists information about the thymocyte pro-apoptotic role for most of the galectins produced by tumors (galectins-1, -3, -8, and -9).
Physiologically, galectin-1 is detected in thymic epithelial, endothelial, and dendritic cells, as well as macrophages [134,135]. From a functional point of view, galectin-1 induces thymocyte apoptosis [136]. This initial result was extensively confirmed under different experimental conditions, allowing a better understanding of this biological phenomenon. The highly proliferating immature CD4+ CD8+ double-positive thymocytes are the primary targets for galectin-1-induced cell death [136,137]. This FAS-independent apoptosis relies on a thymocyte permissive glycophenotype and involves the lectin interaction with CD7, CD43, and CD45 receptors [55,56,58,59,138]. Pro-apoptotic properties of galectin-1 on thymocytes have usually been evaluated in vitro using a soluble recombinant protein; only a few studies have used a more relevant biological context, demonstrating that galectin-1 produced in situ by cells have such a pro-apoptotic effect on T cell lines [46,139,140]. Furthermore, no report has addressed this issue using primary T cells. This point is crucial since galectin-1 is an inactive monomer that becomes a biologically active homodimer through non-covalent bonds with a Kd around 7 μM (equivalent to a concentration of 98 μg/mL) [141,142]. Based on this low homodimerization constant, high amounts of the protein are needed to reach the critical concentration required for the active dimer formation. Even if a genetically engineered dimeric galectin-1 reduces tenfold the concentrations of the protein to achieve a biologically active form [143], the required concentration of the lectin remains high.
Galectin-3 was also detected in epithelial and phagocytic cells in the medulla and, to a lesser extent, in the cortical regions of the thymus [74,144]. It is important to remark that galectin-3 has opposite effects on cells depending on its extracellular or intracellular localization [145,146]. Similar to galectin-1, extracellular galectin-3 induces thymocyte apoptosis. However, galectin-3 preferentially targets a different cell subpopulation (CD4- CD8- double-negative thymocytes) [74]. Furthermore, and although both galectin-1 and galectin-3 induced apoptosis are carbohydrate-dependent, galectin-3-mediated effects are different in several aspects: galectin-3 uses distinct sets of glyco-receptors (does not require CD7) and involves different molecular mechanisms [74]. Further, galectin-3 is more potent at inducing cell apoptosis when compared to galectin-1. However, galectin-3’s pro-apoptotic effects still require concentrations in the order of μM. In addition, this pro-apoptotic effect of extracellular galectin-3 opposes the anti-apoptotic function of intracellular galectin-3 [147]. Additionally, intracellular galectin-3 blocks galectin-1-mediated apoptosis [138], implying that both galectin members are closely interrelated in the control of thymocyte apoptosis.
Galectin-9 induces carbohydrate-dependent cell death in thymocytes [138]. Galectin-9 is detected in epithelial cells throughout the thymus, but it is more abundantly found in the medulla compared to the cortical regions of the thymus [138]. Again, galectin-9 has its particularities when compared against other galectins. Galectin-9 induces the cell death of all thymic subpopulations [138]; other galectins show more population-specific effects. Thymocytes’ apoptosis induced by galectin-9 involves receptors that are different from those used by galectins-1 and -3: while at present the relevant receptors remain unknown, CD44 could be a potential candidate since it has been demonstrated to bind galectin-9 in peripheral T cells [112,113]. At a mechanistic level, galectin-9-mediated apoptosis involves, at least partially, a Bcl-2-mediated pathway [138]. In addition, galectin-9 is more potent than the other galectins at inducing T cell death (<1 μM is effective) [138,148].
Galectin-8 is also found in the thymus but, in contrast to galectins-1, -3, and -9, it is not detected in thymic epithelial cells [149]. This galectin induces apoptosis of CD4+ CD8+ double-positive thymocytes through a mechanism that, at least partially, involves activation of the caspase-mediated pathway. In this in vitro study, concentrations of galectin-8 ranging from 0.5 to 2 μM were effective at inducing apoptosis [149].
Former evidence supports galectins acting as pro-apoptotic factors for thymocytes when produced in situ under physiological situations. Thus, galectins produced abundantly by tumors could shape the repertoire of newly generated T lymphocytes. As previously stated, galectins can circulate through biological fluids and reach the thymus. Although it is difficult to transfer in vitro concentrations to tissue levels, comparing the concentrations of circulating galectins in sera (in the order of ng/mL, as found in the 55 reports currently available for different cancers; some were cited before) with the concentrations of galectins required to trigger thymocyte apoptosis (in the order of μg/mL), the galectin concentrations reaching the thymus are likely insufficient to induce the thymocytes’ cell death. The only way tumor-derived galectins could induce thymocyte apoptosis would be by trapping these lectins, which would allow reaching the required galectin concentrations locally. To date, this phenomenon has not been described. Otherwise, if μM concentrations are reached in biological fluids, galectins may induce dangerous side effects, such as the aggregation of different types of cells [143,150] and potential systemic immunosuppression. Taking these arguments together, it seems unlikely that tumor-derived, circulating galectins can induce cell apoptosis in the thymus.
Apart from apoptosis, other biological properties, such as cell-to-cell interactions, can be regulated by galectins in the thymus [151]. For instance, galectin-3 was described as a factor promoting thymocytes’ release from thymic epithelial cells. Therefore this protein is a de-adhesive factor [144]. Conversely, a pro-adhesive role has been ascribed to galectin-1 through its interaction with several proteins of the extracellular matrix [134]. Thymic galectin-9 also acts as an adhesive molecule since it induces thymocyte homotypic aggregation [150]. Once again, all these biological aspects of galectins have essentially been addressed in vitro and require the use of high concentrations of recombinant proteins. It is, therefore, unlikely that any cell attachment-related modulation in the thymus can be ascribed to circulating and tumor-derived galectins.
Despite former arguments against the role of circulating and tumor-derived galectins in controlling thymic apoptosis and cell attachment-related functions, the evaluation of murine galectin-deficient models highlight that these proteins play essential roles in the control of thymocyte development. Indeed, while data identify endogenous galectin-1 as being not essential for thymocyte development (unaffected general thymic subpopulation numbers and percentages), this protein shapes the T cell repertoire acting as a selective modifier of positive and negative selection processes [152]. Indeed, physiological thymic galectin-1 opposes positive selection while it promotes negative selection of conventional CD8+ T cells. This conclusion was further confirmed by the use of a galectin-1-specific inhibitor in fetal thymic organ cultures; such treatment enhanced CD8+ T cell development [152]. The failures in thymic selection processes observed in galectin-1-deficient mice could explain (at least partially) some of the peripheral autoimmune phenomena spontaneously observed in these mice [153,154].
Endogenous galectin-3 is another member with an important modulation role in thymocyte development. Indeed, thymocyte subpopulations in galectin-3-deficient mice show a reduction in thymus cellularity, which is due to reduced in vivo proliferation and increased apoptosis of thymocytes [155]. Interestingly, although exogenous galectin-3 induces T cell apoptosis, the absence of endogenous galectin-3 also induces thymocyte apoptosis. This last point was demonstrated by genetic ablation and confirmed by specific inhibition experiments. Therefore, it is evident that optimal production of T lymphocytes requires physiological levels of galectin-3.
Up to mid-2021, no report indicates alterations in the thymic selection processes due to gene invalidation of galectins-7, -8, and -9. Nevertheless, it is interesting to note that galectin-7 is detected in Hassal corpuscules located in the medulla of the thymus [156]. While a functional significance remains unclear, this observation indicates a potential role of galectin-7 in thymic regulation. Altogether, data on galectins-1 and -3 supports that serum concentrations of galectins detected in cancer patients could modify the repertoire of T lymphocytes exported from the thymus to the periphery. Mechanisms by which this happens remain to be determined and more studies on the role of other galectins are necessary. Here, it is also important to note that, in addition to their lectin functions that require high concentrations to induce glycan–lectin lattices, galectins also act as regulators of gene expression, transcript maturation, and intracellular signaling [4,68,83,91,93,94,96,157,158,159,160,161]. Most of these later functions are glycan-independent and therefore require lower galectin levels [161]. The galectin concentrations detected in the circulation of cancer patients are consistent with these non-lectin functions. Therefore, galectins can act like soluble factors controlling the thymocyte educative process through effects on the thymocytes themselves or, indirectly, via effects on the stromal cells that participate in their education. Additional basic research can shed some light on these concerns.
1.2. T Lymphocyte Regulation by Galectins at the Periphery
In humans, the peripheral T cell pool is constituted by approximately 4 × 1011 naïve T lymphocytes [123]. In general, analysis of the murine peripheral T cell repertoire has demonstrated that each naïve clonotype is made up of few cells [162]. Similar low frequencies for each T clonotype have been found in humans [163]. More stringent restrictions apply to the anti-tumor-specific T cell repertoire since potent central selection processes only allow the exit of low avidity clonotypes against self-epitopes [164]. On the other hand, the frequency of anti-tumor T cells is increased through the clonotypes reactive against the neoepitopes, which arise as a consequence of tumor genetic instability; these clonotypes are not eliminated by central tolerance [165,166]. In general, basic studies have reported variable, low frequencies of pre-existing anti-tumor T lymphocytes in healthy individuals [167,168,169]. These low numbers of cells constitute the available army to recognize and eliminate transformed cells. In cancer patients, the said T cells expand and gain cytotoxic function in a tumor-dependent manner since each of these cancers differs in their mutational ratio, in their immunogenicity [170], and use of evasion pathways. Despite these considerations, the low number of specific anti-tumor T cells represents a real challenge for immuno-oncology. This fact implies that the expansion and gain of function of the few tumor-specific T cells must be as efficient as possible, at the risk of losing these effector cells. Hence, understanding the tumoral strategies leading to lymphocyte de-functionalization is essential to counteract them during cancer immunotherapies.
To assess the role of tumor-derived galectins on the function of immune cells in the periphery, we will focus on two anatomical locations where tumors have a direct and significant influence: the draining lymph nodes and the tumor itself. It does not imply that circulating galectins (produced by tumors) can influence cellular functions in other anatomical locations; these are beyond the scope of this review.
1.2.1. Galectins’ Functions in Tumor-Draining Lymph Nodes
The first stages of lymphocyte activation occur in the draining node. There, a specific clonal expansion process is carried out [171]. Tumor-derived galectins can reach this anatomical location via blood and lymphatic vessels as soluble proteins, transported by cells or contained in exoparticles [172]. Once in lymph nodes, galectins impact on the early lymphocyte activation process. As mentioned above, there is abundant literature about the pro-apoptotic functions on recently activated T lymphocytes for the 5 galectins evaluated in this review. However, the concentrations of galectins found in blood and lymphatic fluids are unlikely to induce the glycolattice formation required for the said function. As previously discussed in the thymus section, it is therefore unlikely that the tumor-derived galectins in circulation modulate lymphocyte function in lymph nodes through their lectin properties. In contrast, other lymphocyte functions may be finely regulated by circulating tumor-derived galectins.
The emergence of a tumor is associated with a complete reorganization of the local tissue architecture, with major impacts on blood and lymphatic vessels. Indeed, circulating galectin-1 can be taken up by and control the functional properties of endothelial cells [20]. It is worth noting that galectin-1 expressed by endothelial cells plays a major regulatory role in the homing of naïve lymphocytes towards the lymph nodes [135,173,174]. Indeed, lymphocyte recruitment is significantly reduced in vitro. This phenomenon happens when endothelial cells are treated with recombinant galectin-1 at nM concentrations [173] or when endothelial cells upregulate galectin-1 following their incubation with tumor cell-conditioned media [135]. The latter is an example that mimics how tumor products can alter physiology, even at a distance. It is important to note that these nM concentrations (around 14 ng/mL) are compatible with the levels of galectins detected in biological fluids. More importantly, lymphocyte homing is significantly increased in galectin-1 deficient compared to wild-type mice [173]. This biological effect occurs independently of cell death [135] and both in physiological and inflammatory conditions [173]. Thus, tumor-derived galectin-1 decreases the influx of naïve T cells into the draining lymph nodes, accounting for a reduction in T cell activation and clonal expansion.
Despite regulation of cell migration through the blood endothelium, galectin-1 also plays a significant role in the formation of new lymphatic vessels. Indeed, genome-wide functional analysis revealed that galectin-1 is one of the major regulators of lymphatic endothelial cell function [175]. Therefore, this protein has a major impact on how tumor-derived antigens and antigen-presenting cells arrived at the draining lymph node through lymphatic vessels. Furthermore, galectin-1 inhibits the migration of immunogenic dendritic cells through the extracellular matrix and across lymphatic endothelial cells [176].
Galectin-1 compromises cell migration, and the T lymphocytes that effectively reach the draining lymph node are poorly activated if this lectin is present in the local media. Indeed, galectin-1 imparts a regulatory program in dendritic cells, resulting in lower lymphocyte priming [177,178]. However, regulation of the dendritic cell properties by galectin-1 does not exclusively depend on the extracellular concentrations of this lectin since endogenous galectin-1 also controls dendritic cell immunogenic potential [177,179,180]. Altogether, galectin-1 plays an essential role in controlling the initial steps of antigen-specific lymphocyte activation. Indeed, lymphocytes from the draining lymph nodes of galectin-1-silenced tumors are more prone to proliferation and produce higher levels of IL-2 and IFNγ [181,182]. The tumor origin of galectin-1 that causes alterations in antigen presentation is further supported by the fact that these biological effects are observed in the draining but not in other tumor-distant lymph nodes [181].
However, comprehension of the scenario in its full complexity requires additional clues. Indeed, the use of antigen-presenting cell-free systems demonstrated that galectin-1 modulates TCR-mediated signaling [137,183]. Accordingly, galectin-1 directly affects T cells during the early steps of activation, which are not only dependent on accessory cells. Altogether, tumor-derived galectin-1 promotes lymphocyte differentiation towards Th profiles that are inefficient to eliminate transformed cells [28,52,184,185,186]. In this respect, endogenous lymphocyte galectin-1 can control cell function at the level of gene expression regulation (reviewed in [4]). Indeed, endogenous galectin-1 in lymphocytes controls their expansion [187] and differentiation [188,189] in a variety of experimental models. In cancer, our group demonstrated that the inactivation of endogenous galectin-1 in lymphocytes reverses tumor immunosuppression [190].
Finally, tumor-derived galectins participate in the recruitment of cells with regulatory function in lymph nodes and thus have a major impact on the clonal expansion of anti-tumor T lymphocytes. Indeed, galectin-1 silencing in tumors reduces the frequency and the suppressive function of CD4+ CD25+ FOXP3+ regulatory T cells (Tregs) in draining lymph nodes [191]. Furthermore, Tregs require galectin-1 to be fully suppressive; galectin-1 neutralization reverses immunosuppression by Tregs [62,192]. Galectin-1 plays also an important role in the differentiation and suppressive function of CD122+ PD-1+ CD8+ Tregs [193]. In addition, this galectin also attracts other regulatory cells, such as M2 macrophages and myeloid-derived suppressor cells, to the tumor-draining lymph nodes, as it does towards the tumors themselves [194,195,196,197,198].
Galectin-3 is another member of this lectin family with a significant impact on the anti-tumor lymphocyte activation occurring in the draining lymph nodes. Our laboratory recently demonstrated that tumor galectin-3 is a potent negative checkpoint that suppresses lymphocyte proliferation in a prostate cancer microenvironment [199]. Furthermore, galectin-3 downregulation is a pre-requisite for optimal lymphocyte activation when a dendritic cell-based vaccine is used in prostate cancer. In such a case, long-term protective immunity is achieved [199]. Additional evidence suggests that galectin-3 could also act as a negative immune checkpoint in other types of cancers [72,75,79,200].
Among the molecular mechanisms accounting for the powerful lymphocyte inhibitory effect of galectin-3 in cancers, this protein modulates the interactions between T cells and antigen-presenting cells [85]. First, galectin-3 deficient immature dendritic cells have defective motility properties [201]. Consequently, by controlling the dendritic cell migration from the peripheral tissues (including tumors) to the draining lymph nodes, galectin-3 has a direct role in eliciting anti-tumor immune responses. Furthermore, this particular galectin also contributes to dendritic cell homeostasis since it was observed that galectin-3-deficient mice have increased numbers of plasmacytoid dendritic cells [79]. Interestingly, plasmacytoid dendritic cells are superior to conventional ones in activating anti-tumor CD8+ T lymphocytes [79]. Finally, information obtained from experimental models of infection has demonstrated the critical function of galectin-3 on the adaptive immune responses triggered by dendritic cells [202,203,204]. Altogether, these data seem to indicate galectin-3 plays a role at the initial steps of tumor antigen presentation.
Galectin-3 also has a direct effect on T lymphocytes. First, the galectin-3 expression on tumor cells negatively impacts the T lymphocyte numbers in lymph nodes [199]. This effect can be explained through the regulation at the initial steps of the lymphocyte activation process. Indeed, galectin-3 modulates the immunological synapse formation, restricting TCR movements, potentiating TCR downregulation, suppressing early TCR signaling pathways, and controlling cytokine production [73,76,85,205]. Extracellular galectin-3 accomplishes these biological effects via interactions with membrane glyco-receptors as well as by reducing the availability of soluble proteins (in particular cytokines like IL-2, IFNγ, and IL-12) [72]. These functions are glycan-dependent. Subject to the surrounding microenvironment, endogenous galectin-3 is upregulated in T cells early upon activation and skews their differentiation program. Indeed, galectin-3 deficiency promotes immune responses that favor effectors and effector memory T cells to the detriment of the generation of central memory T cells [85,206]. Furthermore, it was described that the survival of recently activated T cells might be affected by galectin-3. Indeed, in vitro studies have demonstrated that extracellular galectin-3 induces apoptosis in human T cells by directly binding the glycoprotein receptors CD45 and CD71 [74]. However, similarly to what was discussed on galectin-1, it is unlikely that tumor-derived circulating galectin-3 reaches the concentrations required to reveal its pro-apoptotic properties in the tumor-draining lymph nodes in cancer patients. It is also important to note that during activation, lymphocytes upregulate intracellular galectin-3 [206,207], which protects T cells from apoptosis [147,206]. Thus, the role of galectin-3 on the survival of newly activated lymphocytes in the lymph nodes is complex, and its real pathologic relevance remains controversial. On the other hand, the expression of galectin-3 by the stroma is required to recruit CD4+ CD25+ FOXP3+ Tregs towards immune organs in tumor-harboring mice [200]. Considering all these arguments, tumor-derived galectin-3 may substantially impact T cell activation, expansion, and polarization of the immune responses elicited in tumor-draining lymph nodes. This concept is relevant not only to the design of in vivo vaccine strategies [199] but also to adoptive T cell transfer of ex vivo-expanded tumor-reactive T cells [208].
Currently, few studies have evaluated the effect of galectin-8 in the process of anti-tumor immune activation. First, galectin-8 crosstalk among the VEFG-C, podoplanin, and integrin pathways plays a key role in lymphangiogenesis [209]. Indeed, podoplanin-expressing macrophages promote lymphangiogenesis in breast cancer via interaction with galectin-8 on lymphatic endothelial cells [210]. Some additional information can be drawn from other (non-tumoral) experimental models. Indeed, galectin-8 promotes all steps of antigen presentation from antigen binding, internalization, processing [211], and maturation of dendritic cells [212,213]. Studies with galectin-8 deficient antigen-presenting cells confirmed the relevance of such functions in pathophysiology [212]. This experimental model seems more relevant compared to artificial in vitro use of high concentrations of recombinant galectin-8. Aside from the antigen-presenting cell-dependent naïve CD4+ T cell co-stimulation that occurs with low galectin-8 concentrations, it was demonstrated that higher concentrations of galectin-8 induce antigen-independent proliferation of CD4+ T cells [214]. However, higher concentrations seem unlikely to be reached in tumor-draining lymph nodes, while low ones could play a role in controlling tumor antigen presentation in those immune organs.
Finally, recombinant galectin-8 increases differentiation of CTLA-4+ IL-10+ CD103+ Tregs through activation of TGF-β and sustained-IL-2 receptor signaling [215]. Tumors could use this strategy to block immunity in draining lymph nodes. In summary, little is known about the biological functions of galectin-8 in lymph nodes during cancer. Compilation of existing data indicates that secretion of this protein would not generate a strong selective advantage for tumors. On the contrary, galectin-8-based strategies could potentiate anti-tumor immunity since this lectin can lower the TCR activation threshold [216].