HCV genomic RNA replication occurs in the replication organelles (RO) and is tightly linked to ER membrane alterations containing replication complexes (proteins NS3 to NS5B). The amplification of HCV genomic RNA could be regulated by the RO biogenesis, the viral RNA structure (i.e., cis-acting replication elements), and both viral and cellular proteins. Studies on HCV replication have led to the development of direct-acting antivirals (DAAs) targeting the replication complex.
- hepatitis C virus
- replication organelles
- NS3 to NS5B proteins
1. Introduction
Infection with the hepatitis C virus (HCV) can cause chronic hepatitis C (CHC), liver cirrhosis, hepatocellular carcinoma, and other extra-hepatic manifestations. The prevalence of CHC patients worldwide was around 71 million in 2017 (https://www.who.int/hepatitis/publications/global-hepatitis-report2017/en/). HCV belongs to the family Flaviviridae and genus Hepacivirus. Its genome is a single-stranded RNA with positive polarity. Many different but closely related circulating HCV variants (i.e., quasispecies) can be detected in CHC patients due to the low fidelity of the HCV RNA polymerase (NS5B) and its high replication rate [1]. Thus, HCV genomic RNA sequences are highly heterogeneous among different isolates. At present, HCV is classified into at least six major genotypes (GT 1 to 6) [2][3]. The geographic distribution of different HCV genotypes varies [3]. Subtype 1a is found throughout the US and Northern Europe, while subtype 1b is widely distributed throughout the world and is a major subtype in Japan. Genotype 2 is present in the same areas as genotype 1. Subtype 3a is widely distributed in South Asia and Oceania, while subtype 3b is mainly found in East Asia. Genotype 4 is mainly present in the Middle East, Northern to Central Africa, and Europe. Subtype 5a is mainly found in South Africa. Genotype 6 is mainly distributed throughout East and South-East Asia.
The life cycle of HCV begins with its binding to cells. Numerous cellular factors, including proteins, lipids, and glycans, promote the entry of HCV particles into hepatocytes. HCV initially attaches to the surface proteoglycans, e.g., the scavenger receptor BI, and to the tetraspanin CD81. After lateral translocation to tight junctions, claudin-1 and occludin proteins become essential for HCV entry. HCV particles are engulfed by clathrin-mediated endocytosis and then fused with endosomal membranes in low-pH conditions. Viral genomic RNA is then released into the cytoplasm [4]. Then, the HCV genomic RNA is used for both protein translation and viral RNA replication. HCV RNA replication takes place within the replication organelles (RO) in the endoplasmic reticulum (ER). Finally, HCV utilizes the biosynthetic pathway of very-low-density lipoprotein to assemble the viral particles and egress from the cells [5].
The HCV RNA genome (~9600 nucleotides) possesses one open reading frame that is flanked by 5’ and 3’ untranslated regions (UTRs) (Figure 1a). Translation of the viral RNA leads to the synthesis of a polyprotein, which is processed into individual viral proteins via cleavages of both cellular and viral proteases. The structural proteins (i.e., the core and envelope glycoproteins E1 and E2) are the main constituents of HCV particles, whereas the viroporin p7 and nonstructural protein 2 (NS2) are involved in virion assembly [6]. The remaining nonstructural proteins (i.e., NS3, NS4A, NS4B, NS5A, and NS5B; NS3-NS5B) that have specific roles in viral genome amplification form the replication complex [7][8][9]. The roles of different viral proteins in HCV replication are summarized in Table 1.
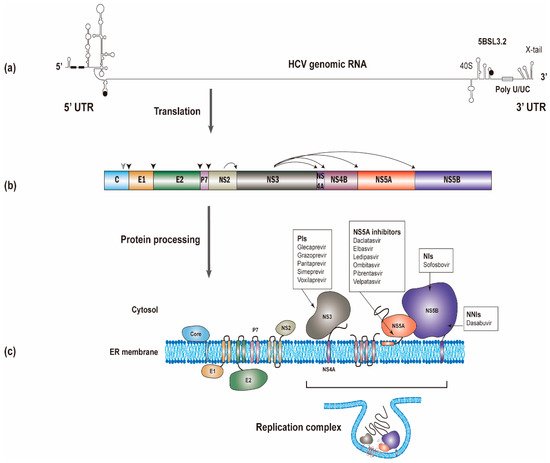
Figure 1. The synthesis of the hepatitis C virus (HCV) proteins. (a) The start and stop codons for protein translation were marked by black circles, while two recognition sites on the 5’ UTR for miR-122 were marked by black rectangles. (b) The polyprotein is co- and post-translationally cleaved by cellular or viral proteases to yield the structural proteins (core, E1 and E2) and the nonstructural proteins (p7, NS2, NS3, NS4A, NS4B, NS5A and NS5B proteins). The core, E1, and E2 are processed by cellular signal peptidase (filled arrowhead). A mature core protein will be generated after further cleavage by signal peptide peptidase (empty arrowhead). The NS2/NS3 junction site is cleaved by the NS2-NS3 auto-protease, and the remaining nonstructural proteins are processed by the NS3/4A proteinase. (c) All of the HCV proteins are directly or indirectly associated with the endoplasmic reticulum. Currently used anti-HCV direct-acting antivirals (DAAs) target NS3, NS5A, and NS5B, respectively. NS3, NS4A, NS4B, NS5A, and NS5B proteins will form the replication complex.
Table 1. Hepatitis C virus (HCV) proteins play different roles in viral replication.
| Viral Protein | Role in HCV Replication |
|---|---|
| Core | Package HCV genomic RNA to form nucleocapsids and also involve in lipid synthesis |
| E1, E2 | Responsible for the entry of virions to cells |
| p7 | Ion channel |
| NS2 | Auto-protease to cleave the junction between NS2 and NS3 |
| NS3 | NS3 contains an amino-terminal protease domain responsible for the HCV polyprotein processing and a carboxy-terminal DExD-box helicase domain responsible for HCV RNA replication through unwinding RNA secondary structures |
| NS4A | Cofactor for NS3 protease |
| NS4B | To serve as a scaffold for the viral replication complex and to induce the rearrangements of membrane vesicles |
| NS5A | To interact with a large number of cellular proteins that are important for viral assembly and function of the replication complex |
| NS5B | HCV RNA-dependent–RNA-polymerase responsible for HCV RNA amplification |
Over thirty years of research on the mechanisms of HCV replication has led to the successful development of direct-acting antivirals (DAAs) targeting the replication complex [10].
2. Viral Replication Organelles (RO)
HCV induces cellular membrane alterations referred to as the membrane web (MW) for viral RNA replication [11]. Different types of membrane alterations induced by HCV were observed [12][13]. Among these membrane alterations, double-membrane vesicles (DMVs) induced by HCV infection associated with double-stranded RNA (dsRNA) and nonstructural proteins are believed to be the sites of viral genome replication (i.e., viral replication organelles (RO)) in cultured cells (Figure 2). DMVs comprise the predominant HCV-induced membrane structure that forms in the cytoplasm close to the lipid droplets (LDs) in cultured cells. LDs with HCV core and NS5A proteins surrounded by ER is close to the HCV replication (e.g., DMV) and assembly sites. HCV genomic RNA synthesized in the DMVs is transferred by HCV nonstructural proteins and encapsidated by the core proteins to form the nucleocapsid. The HCV nucleocapsid will then interact with glycoproteins E1/E2 in the assembly sites and bud into the ER lumen [14]. DMVs are heterogeneous in size, with an average diameter of ~200 nm. At late time points after infection, multi-membrane vesicles were observed and believed to reflect a stress response induced by high-level virus replication [13][15][16]. These HCV-induced DMVs are morphologically similar to those identified in cells infected with coronaviruses, picornaviruses and noroviruses [17]. Previous studies also showed that HCV could induce membrane alterations in the hepatocytes of HCV-infected patients [18][19].
HCV-induced single-membrane vesicles (SMVs) were also detected sporadically in cultured cells [13][15][16]. Unlike observations from the cultured cells, a recent report showed that the MW detected in liver tissues of HCV-infected patients seems essentially to be made of clusters of SMVs [20]. Further studies are needed to clarify this issue.
The majority of HCV DMVs appear to be closed structures, and only a few of them have an opening pore toward the cytosol [13]. It is not yet known whether HCV RNA replication takes place on the interior or exterior membrane surface of the DMVs. If HCV RNA replication occurs on the interior surface of DMVs, then a transport mechanism must be present to allow the influx of metabolites (e.g., nucleoside triphosphates) required for replication and the exit of newly synthesized viral RNAs for translation or virion assembly [8]. This hypothesis is supported by the findings that HCV hijacks specific cellular components responsible for nucleocytoplasmic transport and that these cellular factors are probably involved in maintaining a transport system between the cytosol and the interior of viral ROs [21][22].
There are several advantages to forming viral ROs for HCV RNA synthesis [17]. First, the viral replication complex (NS3-NS5B) and cellular factors responsible for HCV RNA replication can be concentrated in ROs. Second, ROs, by excluding cellular RNAs, contribute to the template specificity of the replication complex. Third, the replication intermediates (i.e., dsRNA) can be protected from the detection of cellular innate immune sensors. Fourth, ROs facilitate the separation of different stages in the life cycle of HCV (translation vs. replication, replication vs. assembly) by compartmentalization [22]. Fifth, several reports showed that viral RNA and proteins associated with the viral ROs are protected from cellular proteases and nucleases, indicating that RNA replication occurs in a membranous environment separated from the surrounding cytoplasm [16][23][24].
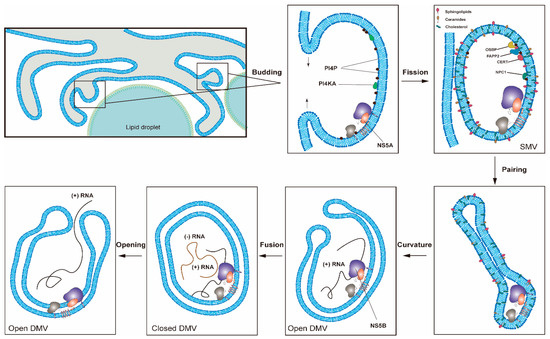
Figure 2. One proposed model for the formation of HCV replication organelles (RO). Double-membrane vesicles (DMV) biogenesis is a complex process possibly requiring several membrane remodeling steps, including budding, fission, pairing, curvature, and fusion [11][17]. HCV DMV formation can be induced by NS5A with the help of other nonstructural proteins [25][26]. HCV activates the lipid kinase PI4KIIIα to generate enhanced levels of PI4P, which in turn attracts lipid transport proteins (e.g., OSBP, FAPP2, NPC1 and CERT) delivering cholesterol and glycosphingolipids into DMVs. PSTPIP2, a protein with membrane-deforming activity, and PLA2G4C are also critical for membrane web (MW) formation. HCV RNA replication can be conducted by NS5B with the help of other nonstructural proteins in the closed DMVs. After the completion of HCV genomic RNA synthesis, the newly synthesized viral RNAs will be released from the open DMVs (possibly with Kaps or Nups) for translation or virion assembly. Other possible models for DMV formation have also been proposed [11].
2.1. HCV Proteins Involved in RO Formation
Although any protein of the HCV replication complex (NS3-NS5B) can induce membrane alterations, NS5A is the only one capable of inducing DMV formation [13][25][26]. The insertion of the amino-terminal amphipathic α-helix of NS5A into just one membrane layer could facilitate the membrane curvature required for RO formations [27]. The observation that NS5A inhibitors (e.g., daclatasvir) block the HCV RO formation independent of RNA replication demonstrated the essential role of NS5A in the RO formation [28]. The efficiency of the DMV formation induced by NS5A alone is low but is greatly enhanced when the other nonstructural proteins are also expressed, e.g., NS4B, NS3, or NS5B [26][29][30].
Similar to NS5A, NS4B also contains terminal amphipathic α-helices, which can alter membrane properties. Moreover, the membrane topology of NS4B is likely to undergo posttranslational changes [31][32][33][34], possibly in an NS5A-regulated manner [35]. Moreover, NS4B contains a GXXXXGK P-loop for nucleotide triphosphate binding, which may be involved in membrane rearrangements [36]. In addition, NS4B could form homo-oligomeric complexes, which are required for the RO formation [29][37][38].
2.2. Cellular Factors Involved in HCV RO Formation
In addition to having direct involvement in the RO formation, viral proteins also contribute to membrane alterations by recruiting cellular factors required for RO biogenesis. For example, NS5A. Cyclophilin A (CypA), receptor for activated protein C kinase 1 (RACK1) and ATG14L were found to participate in DMV formation for HCV replication by interacting with NS5A [39][40][41][42], while Surf4 and prolactin regulatory element-binding (PREB) participated by interacting with NS4B [43][44]. PSTPIP2 (Proline-serine-threonine phosphatase interacting protein 2) with membrane-deforming activity and PLA2G4C (cytosolic phospholipase A2 gamma) are also important for HCV RO formation via direct interactions with NS4B and NS5A [45][46].
The HCV replication complex is reportedly associated with membrane lipid micro-domains (i.e., lipid rafts) [47][48], enriched with cholesterol, sphingolipids, and certain proteins. Lipid rafts generally contain three to five times the cholesterol content found in the surrounding bilayer [49]. Shaping an ER membrane into an RO for HCV RNA replication requires not only viral and cellular proteins but also lipid synthesis [50][51][52]. Multiple reports have indicated that HCV modulates lipid metabolism (e.g., cholesterol and fatty acid biosynthesis) to promote viral replication [53][54][55]. This modulation results in de novo lipid biosynthesis in order to increase the membrane surface area required for the RO formation. SREBPs (the sterol regulatory element-binding protein) are major regulators of lipid metabolism and major transcription factors for the expression of genes required for lipid biosynthesis [56]. HCV NS4B has been shown to activate SREBP, leading to the elevated transcription of genes involved in lipogenesis, e.g., fatty acid synthase (FASN) [57].
Modulation of the lipid environment of RO via HCV also includes the recruitment and activation of the lipid kinase PI4KIIIα by NS5A and NS5B proteins to generate enhanced levels of phosphatidylinositol 4-phosphate (PI4P) at the RO [58]. PI4P could attract lipid transport proteins (oxysterol-binding protein (OSBP), four-phosphate adaptor protein 2 (FAPP2), NPC1, and ceramide transfer protein (CERT) to deliver glycosphingolipids, cholesterol, and ceramide to RO, respectively [16][59][60]. Recently, it was reported that HCV NS3/4A protease controls the activity of 24-dehydrocholesterol reductase (DHCR24), catalyzing the conversion of desmosterol to cholesterol and regulating the lipid environment for HCV RNA replication [61]. In contrast, cholesterol-25-hydroxylase induced by interferon could block MW formation via the production of 25-hydroxycholesterol and thus restrict HCV replication [62]. Recently, C19orf66, an interferon-stimulated gene, was reported to inhibit HCV by preventing the elevation of PI4P and altering RO formation [63].
In addition to these cellular factors, several studies have shown that autophagy plays an early role in establishing HCV replication [64][65][66]. DMVs induced by HCV accumulated at the MW are morphologically similar to autophagosomes [15]. Thus, autophagy may help to induce MW formation during HCV replication [67]. However, DMVs induced by HCV with an average diameter of ∼200 nm are smaller than autophagosomes of 500 to 1000 nm in diameter. The exact role of autophagy in HCV RO formation requires further investigation [68].
Proteins in the nuclear transport machinery (including soluble nuclear transport factors (NTFs), e.g., karyopherins (Kaps)) and nucleoporins (Nups) in the nuclear pore complexes (NPCs) are probably involved in the transfer between the cytosol and the viral ROs [21][22].
References
- Martell, M.; Esteban, J.I.; Quer, J.; Genesca, J.; Weiner, A.; Esteban, R.; Guardia, J.; Gomez, J. Hepatitis C virus (HCV) circulates as a population of different but closely related genomes: Quasispecies nature of HCV genome distribution. J. Virol. 1992, 66, 3225–3229.
- Li, H.C.; Lo, S.Y. Hepatitis C virus: Virology, diagnosis and treatment. World J. Hepatol. 2015, 7, 1377–1389.
- Shin-I, T.; Sugiyama, M.; Mizokami, M. Hepatitis C Virus Genotypes and Their Evolution. In Hepatitis C Virus I: Cellular and Molecular Virology; Miyamura, T.L., Lemon, S.M., Walker, C.M., Wakita, T., Eds.; Springer: Tokyo, Japan, 2016; pp. 24–29.
- Colpitts, C.C.; Tsai, P.L.; Zeisel, M.B. Hepatitis C Virus Entry: An Intriguingly Complex and Highly Regulated Process. Int. J. Mol. Sci. 2020, 21, 2091.
- Alazard-Dany, N.; Denolly, S.; Boson, B.; Cosset, F.L. Overview of HCV Life Cycle with a Special Focus on Current and Possible Future Antiviral Targets. Viruses 2019, 11, 30.
- Moriishi, K.M.; Matsuura, Y. Structural Proteins of HCV and Biological Functions. In Hepatitis C Virus I: Cellular and Molecular Virology; Miyamura, T.L., Lemon, S.M., Walker, C.M., Wakita, T., Eds.; Springer: Tokyo, Japan, 2016; pp. 105–127.
- Tabata, K.; Neufeldt, C.J.; Bartenschlager, R. Hepatitis C Virus Replication. Cold Spring Harb. Perspect. Med. 2020, 10, a037093.
- Suzuki, T. Hepatitis C Virus Replication. Adv. Exp. Med. Biol 2017, 997, 199–209.
- Suzuki, T.; Suzuki, R. Role of Nonstructural Proteins in HCV Replication. In Hepatitis C Virus I: Cellular and Molecular Virology; Miyamura, T.L., Lemon, S.M., Walker, C.M., Wakita, T., Eds.; Springer: Tokyo, Japan, 2016; pp. 129–148.
- Parlati, L.; Hollande, C.; Pol, S. Treatment of hepatitis C virus infection. Clin. Res. Hepatol. Gastroenterol. 2020, 2020, 101578.
- Wang, H.; Tai, A.W. Mechanisms of Cellular Membrane Reorganization to Support Hepatitis C Virus Replication. Viruses 2016, 8, 142.
- Ferraris, P.; Beaumont, E.; Uzbekov, R.; Brand, D.; Gaillard, J.; Blanchard, E.; Roingeard, P. Sequential biogenesis of host cell membrane rearrangements induced by hepatitis C virus infection. Cell. Mol. Life Sci. 2013, 70, 1297–1306.
- Romero-Brey, I.; Merz, A.; Chiramel, A.; Lee, J.Y.; Chlanda, P.; Haselman, U.; Santarella-Mellwig, R.; Habermann, A.; Hoppe, S.; Kallis, S.; et al. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog. 2012, 8, e1003056.
- Lee, J.Y.; Cortese, M.; Haselmann, U.; Tabata, K.; Romero-Brey, I.; Funaya, C.; Schieber, N.L.; Qiang, Y.; Bartenschlager, M.; Kallis, S.; et al. Spatiotemporal Coupling of the Hepatitis C Virus Replication Cycle by Creating a Lipid Droplet-Proximal Membranous Replication Compartment. Cell Rep. 2019, 27, 3602–3617.e5.
- Ferraris, P.; Blanchard, E.; Roingeard, P. Ultrastructural and biochemical analyses of hepatitis C virus-associated host cell membranes. J. Gen. Virol. 2010, 91 Pt 9, 2230–2237.
- Paul, D.; Hoppe, S.; Saher, G.; Krijnse-Locker, J.; Bartenschlager, R. Morphological and biochemical characterization of the membranous hepatitis C virus replication compartment. J. Virol. 2013, 87, 10612–10627.
- Wolff, G.; Melia, C.E.; Snijder, E.J.; Barcena, M. Double-Membrane Vesicles as Platforms for Viral Replication. Trends Microbiol. 2020, 28, 1022–1033.
- Jackson, D.; Tabor, E.; Gerety, R.J. Acute non-A, non-B hepatitis: Specific ultrastructural alterations in endoplasmic reticulum of infected hepatocytes. Lancet 1979, 1, 1249–1250.
- Shimizu, Y.K. Ultrastructural alterations and expression of cytoplasmic antigen 48-1 in hepatocytes in association with hepatitis C virus infection. Microbiol. Immunol. 1992, 36, 911–922.
- Blanchard, E.; Roingeard, P. The Hepatitis C Virus-Induced Membranous Web in Liver Tissue. Cells 2018, 7, 191.
- Neufeldt, C.J.; Joyce, M.A.; Levin, A.; Steenbergen, R.H.; Pang, D.; Shields, J.; Tyrrell, D.L.; Wozniak, R.W. Hepatitis C virus-induced cytoplasmic organelles use the nuclear transport machinery to establish an environment conducive to virus replication. PLoS Pathog. 2013, 9, e1003744.
- Neufeldt, C.J.; Joyce, M.A.; Van Buuren, N.; Levin, A.; Kirkegaard, K.; Gale, M., Jr.; Tyrrell, D.L.; Wozniak, R.W. The Hepatitis C Virus-Induced Membranous Web and Associated Nuclear Transport Machinery Limit Access of Pattern Recognition Receptors to Viral Replication Sites. PLoS Pathog. 2016, 12, e1005428.
- Miyanari, Y.; Hijikata, M.; Yamaji, M.; Hosaka, M.; Takahashi, H.; Shimotohno, K. Hepatitis C virus non-structural proteins in the probable membranous compartment function in viral genome replication. J. Biol. Chem. 2003, 278, 50301–50308.
- Quinkert, D.; Bartenschlager, R.; Lohmann, V. Quantitative analysis of the hepatitis C virus replication complex. J. Virol. 2005, 79, 13594–13605.
- Egger, D.; Wolk, B.; Gosert, R.; Bianchi, L.; Blum, H.E.; Moradpour, D.; Bienz, K. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 2002, 76, 5974–5984.
- Romero-Brey, I.; Berger, C.; Kallis, S.; Kolovou, A.; Paul, D.; Lohmann, V.; Bartenschlager, R. NS5A Domain 1 and Polyprotein Cleavage Kinetics Are Critical for Induction of Double-Membrane Vesicles Associated with Hepatitis C Virus Replication. MBio 2015, 6, e00759.
- McMahon, H.T.; Boucrot, E. Membrane curvature at a glance. J. Cell Sci. 2015, 128, 1065–1070.
- Berger, C.; Romero-Brey, I.; Radujkovic, D.; Terreux, R.; Zayas, M.; Paul, D.; Harak, C.; Hoppe, S.; Gao, M.; Penin, F.; et al. Daclatasvir-like inhibitors of NS5A block early biogenesis of hepatitis C virus-induced membranous replication factories, independent of RNA replication. Gastroenterology 2014, 147, 1094–1105.e25.
- Paul, D.; Madan, V.; Ramirez, O.; Bencun, M.; Stoeck, I.K.; Jirasko, V.; Bartenschlager, R. Glycine Zipper Motifs in Hepatitis C Virus Nonstructural Protein 4B Are Required for the Establishment of Viral Replication Organelles. J. Virol. 2018, 92, e01890-17.
- Paul, D.; Romero-Brey, I.; Gouttenoire, J.; Stoitsova, S.; Krijnse-Locker, J.; Moradpour, D.; Bartenschlager, R. NS4B self-interaction through conserved C-terminal elements is required for the establishment of functional hepatitis C virus replication complexes. J. Virol. 2011, 85, 6963–6976.
- Palomares-Jerez, F.; Nemesio, H.; Villalain, J. The membrane spanning domains of protein NS4B from hepatitis C virus. Biochim. Biophys. Acta 2012, 1818, 2958–2966.
- Palomares-Jerez, M.F.; Nemesio, H.; Franquelim, H.G.; Castanho, M.A.; Villalain, J. N-terminal AH2 segment of protein NS4B from hepatitis C virus. Binding to and interaction with model biomembranes. Biochim. Biophys. Acta 2013, 1828, 1938–1952.
- Palomares-Jerez, M.F.; Nemesio, H.; Villalain, J. Interaction with membranes of the full C-terminal domain of protein NS4B from hepatitis C virus. Biochim. Biophys. Acta 2012, 1818, 2536–2549.
- Ouldali, M.; Moncoq, K.; de la Valette, A.C.; Arteni, A.A.; Betton, J.M.; Lepault, J. Study of membrane deformations induced by Hepatitis C protein NS4B and its terminal amphipathic peptides. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183537.
- Lundin, M.; Lindstrom, H.; Gronwall, C.; Persson, M.A.A. Dual topology of the processed hepatitis C virus protein NS4B is influenced by the NS5A protein. J. Gen. Virol. 2006, 87 Pt 11, 3263–3272.
- Einav, S.; Elazar, M.; Danieli, T.; Glenn, J.S. A nucleotide binding motif in hepatitis C virus (HCV) NS4B mediates HCV RNA replication. J. Virol. 2004, 78, 11288–11295.
- Gouttenoire, J.; Montserret, R.; Paul, D.; Castillo, R.; Meister, S.; Bartenschlager, R.; Penin, F.; Moradpour, D. Aminoterminal amphipathic alpha-helix AH1 of hepatitis C virus nonstructural protein 4B possesses a dual role in RNA replication and virus production. PLoS Pathog. 2014, 10, e1004501.
- Gouttenoire, J.; Roingeard, P.; Penin, F.; Moradpour, D. Amphipathic alpha-helix AH2 is a major determinant for the oligomerization of hepatitis C virus nonstructural protein 4B. J. Virol. 2010, 84, 12529–12537.
- Madan, V.; Paul, D.; Lohmann, V.; Bartenschlager, R. Inhibition of HCV replication by cyclophilin antagonists is linked to replication fitness and occurs by inhibition of membranous web formation. Gastroenterology 2014, 146, 1361–1372.e9.
- Lee, J.S.; Tabata, K.; Twu, W.I.; Rahman, M.S.; Kim, H.S.; Yu, J.B.; Jee, M.H.; Bartenschlager, R.; Jang, S.K. RACK1 mediates rewiring of intracellular networks induced by hepatitis C virus infection. PLoS Pathog. 2019, 15, e1008021.
- Chatterji, U.; Bobardt, M.; Tai, A.; Wood, M.; Gallay, P.A. Cyclophilin and NS5A inhibitors, but not other anti-hepatitis C virus (HCV) agents, preclude HCV-mediated formation of double-membrane-vesicle viral factories. Antimicrob. Agents Chemother. 2015, 59, 2496–2507.
- Chatterji, U.; Bobardt, M.; Schaffer, L.; Wood, M.; Gallay, P.A. Cyclophilin Inhibitors Remodel the Endoplasmic Reticulum of HCV-Infected Cells in a Unique Pattern Rendering Cells Impervious to a Reinfection. PLoS ONE 2016, 11, e0159511.
- Kong, L.; Aoyagi, H.; Yang, Z.; Ouyang, T.; Matsuda, M.; Fujimoto, A.; Watashi, K.; Suzuki, R.; Arita, M.; Yamagoe, S.; et al. Surfeit 4 Contributes to the Replication of Hepatitis C Virus Using Double-Membrane Vesicles. J. Virol. 2020, 94, e00858-19.
- Kong, L.; Fujimoto, A.; Nakamura, M.; Aoyagi, H.; Matsuda, M.; Watashi, K.; Suzuki, R.; Arita, M.; Yamagoe, S.; Dohmae, N.; et al. Prolactin Regulatory Element Binding Protein Is Involved in Hepatitis C Virus Replication by Interaction with NS4B. J. Virol. 2016, 90, 3093–3111.
- Chao, T.C.; Su, W.C.; Huang, J.Y.; Chen, Y.C.; Jeng, K.S.; Wang, H.D.; Lai, M.M. Proline-serine-threonine phosphatase-interacting protein 2 (PSTPIP2), a host membrane-deforming protein, is critical for membranous web formation in hepatitis C virus replication. J. Virol. 2012, 86, 1739–1749.
- Xu, S.; Pei, R.; Guo, M.; Han, Q.; Lai, J.; Wang, Y.; Wu, C.; Zhou, Y.; Lu, M.; Chen, X. Cytosolic phospholipase A2 gamma is involved in hepatitis C virus replication and assembly. J. Virol. 2012, 86, 13025–13037.
- Aizaki, H.; Lee, K.J.; Sung, V.M.; Ishiko, H.; Lai, M.M. Characterization of the hepatitis C virus RNA replication complex associated with lipid rafts. Virology 2004, 324, 450–461.
- Shi, S.T.; Lee, K.J.; Aizaki, H.; Hwang, S.B.; Lai, M.M. Hepatitis C virus RNA replication occurs on a detergent-resistant membrane that cofractionates with caveolin-2. J. Virol. 2003, 77, 4160–4168.
- Anchisi, L.; Dessi, S.; Pani, A.; Mandas, A. Cholesterol homeostasis: A key to prevent or slow down neurodegeneration. Front. Physiol. 2012, 3, 486.
- Alvisi, G.; Madan, V.; Bartenschlager, R. Hepatitis C virus and host cell lipids: An intimate connection. RNA Biol. 2011, 8, 258–269.
- Bley, H.; Schobel, A.; Herker, E. Whole Lotta Lipids-from HCV RNA Replication to the Mature Viral Particle. Int. J. Mol. Sci. 2020, 21, 2888.
- Strating, J.R.; van Kuppeveld, F.J. Viral rewiring of cellular lipid metabolism to create membranous replication compartments. Curr. Opin. Cell Biol. 2017, 47, 24–33.
- Su, A.I.; Pezacki, J.P.; Wodicka, L.; Brideau, A.D.; Supekova, L.; Thimme, R.; Wieland, S.; Bukh, J.; Purcell, R.H.; Schultz, P.G.; et al. Genomic analysis of the host response to hepatitis C virus infection. Proc. Natl. Acad. Sci. USA 2002, 99, 15669–15674.
- Kapadia, S.B.; Chisari, F.V. Hepatitis C virus RNA replication is regulated by host geranylgeranylation and fatty acids. Proc. Natl. Acad. Sci. USA 2005, 102, 2561–2566.
- Diamond, D.L.; Syder, A.J.; Jacobs, J.M.; Sorensen, C.M.; Walters, K.A.; Proll, S.C.; McDermott, J.E.; Gritsenko, M.A.; Zhang, Q.; Zhao, R.; et al. Temporal proteome and lipidome profiles reveal hepatitis C virus-associated reprogramming of hepatocellular metabolism and bioenergetics. PLoS Pathog. 2010, 6, e1000719.
- Waris, G.; Felmlee, D.J.; Negro, F.; Siddiqui, A. Hepatitis C virus induces proteolytic cleavage of sterol regulatory element binding proteins and stimulates their phosphorylation via oxidative stress. J. Virol. 2007, 81, 8122–8130.
- Park, C.Y.; Jun, H.J.; Wakita, T.; Cheong, J.H.; Hwang, S.B. Hepatitis C virus nonstructural 4B protein modulates sterol regulatory element-binding protein signaling via the AKT pathway. J. Biol. Chem. 2009, 284, 9237–9246.
- Reiss, S.; Harak, C.; Romero-Brey, I.; Radujkovic, D.; Klein, R.; Ruggieri, A.; Rebhan, I.; Bartenschlager, R.; Lohmann, V. The lipid kinase phosphatidylinositol-4 kinase III alpha regulates the phosphorylation status of hepatitis C virus NS5A. PLoS Pathog. 2013, 9, e1003359.
- Khan, I.; Katikaneni, D.S.; Han, Q.; Sanchez-Felipe, L.; Hanada, K.; Ambrose, R.L.; Mackenzie, J.M.; Konan, K.V. Modulation of hepatitis C virus genome replication by glycosphingolipids and four-phosphate adaptor protein 2. J. Virol. 2014, 88, 12276–12295.
- Stoeck, I.K.; Lee, J.Y.; Tabata, K.; Romero-Brey, I.; Paul, D.; Schult, P.; Lohmann, V.; Kaderali, L.; Bartenschlager, R. Hepatitis C Virus Replication Depends on Endosomal Cholesterol Homeostasis. J. Virol 2018, 92, e01196-17.
- Tallorin, L.; Villareal, V.A.; Hsia, C.Y.; Rodgers, M.A.; Burri, D.J.; Pfeil, M.P.; Llopis, P.M.; Lindenbach, B.D.; Yang, P.L. Hepatitis C virus NS3-4A protease regulates the lipid environment for RNA replication by cleaving host enzyme 24-dehydrocholesterol reductase. J. Biol. Chem. 2020, 295, 12426–12436.
- Anggakusuma; Romero-Brey, I.; Berger, C.; Colpitts, C.C.; Boldanova, T.; Engelmann, M.; Todt, D.; Perin, P.M.; Behrendt, P.; Vondran, F.W.; et al. Interferon-inducible cholesterol-25-hydroxylase restricts hepatitis C virus replication through blockage of membranous web formation. Hepatology 2015, 62, 702–714.
- Kinast, V.; Plociennikowska, A.; Anggakusuma; Bracht, T.; Todt, D.; Brown, R.J.P.; Boldanova, T.; Zhang, Y.; Bruggemann, Y.; Friesland, M.; et al. C19orf66 is an interferon-induced inhibitor of HCV replication that restricts formation of the viral replication organelle. J. Hepatol. 2020, 73, 549–558.
- Dreux, M.; Chisari, F.V. Autophagy proteins promote hepatitis C virus replication. Autophagy 2009, 5, 1224–1225.
- Dreux, M.; Gastaminza, P.; Wieland, S.F.; Chisari, F.V. The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. USA 2009, 106, 14046–14051.
- Guevin, C.; Manna, D.; Belanger, C.; Konan, K.V.; Mak, P.; Labonte, P. Autophagy protein ATG5 interacts transiently with the hepatitis C virus RNA polymerase (NS5B) early during infection. Virology 2010, 405, 1–7.
- Mohl, B.P.; Bartlett, C.; Mankouri, J.; Harris, M. Early events in the generation of autophagosomes are required for the formation of membrane structures involved in hepatitis C virus genome replication. J. Gen. Virol. 2016, 97, 680–693.
- Chan, S.T.; Ou, J.J. Hepatitis C Virus-Induced Autophagy and Host Innate Immune Response. Viruses 2017, 9, 224.