In physiology and pathophysiology the molecules involved in blood cell–blood cell and blood cell–endothelium interactions have been identified. Platelet aggregation and adhesion to the walls belonging to vessels involve glycoproteins (GP), GP llb and GP llla and the GP Ib–IX–V complex. Red blood cells (RBCs) in normal situations have little interaction with the endothelium. Abnormal adhesion of RBCs was first observed in sickle cell anemia involving vascular cell adhesion molecule (VCAM)-1, α4β1, Lu/BCAM, and intercellular adhesion molecule (ICAM)-4. More recently RBC adhesion was found to be increased in retinal-vein occlusion (RVO) and in polycythemia vera (PV). The molecules which participate in this process are phosphatidylserine and annexin V in RVO, and phosphorylated Lu/BCAM and α5 laminin chain in PV. The additional adhesion in diabetes mellitus occurs due to the glycated RBC band 3 and the advanced glycation end-product receptors. The multiligand receptor binds advanced glycation end products (AGEs) or S100 calgranulins, or β-amyloid peptide. This receptor for advanced glycation end products is known as RAGE. The binding to RAGE-activated endothelial cells leads to an inflammatory reaction and a prothrombotic state via NADPH activation and altered gene expression. RAGE blockade is a potential target for drugs preventing the deleterious consequences of RAGE activation.
- blood cells
- endothelial cells
- thrombosis
- inflammation
- vascular occlusion
1. Introduction
Hemorrhagic syndrome was a major cause of death in prehistoric life until the 19th century. Coagulation was first discovered and the fibrin, which was previously called plasmin, identified in the second part of the 19th century. The role of platelets in the first step of bleeding arrest was only recognized in the first part of the twentieth century. In developed countries, beside wars, thrombosis seems to have been another major cause of death in cardiovascular disorders, coronary syndrome, and cerebral ischemia.
The development of molecular biology and cell biology opened a completely new paradigm in thrombosis and hemostasis. After the discovery of the coagulation factors, the molecules present on blood cells and the vessel walls, endothelial cell smooth muscle cell molecules, and sub-endothelium structures were explored. A new step was made when, from the molecules involved in thrombotic and hemostatic processes, we had access to the gene and the gene regulation. The molecules involved in platelet–vessel wall interactions were detected first. Studies of patients with platelet dysfunction such as Glanzmann thrombasthenia, and Bernard–Soulier syndrome, led to the discovery of the role of glycoproteins (GP IIb/IIIa) in terms of their platelet aggregation and GP Ib–IX–V complex with regard to platelet adhesion.
Hemorheological factors were found to have a major function in blood cells and blood cell–vessel wall interactions. Beside plasmatic factors, blood cells participate in the thrombus formation. The role of platelets was first recognized but with the development of hemorheology, the importance of red blood cells in vessel occlusion and clot formation were investigated. Leukocyte functions were characterized during the 20th century. In inflammatory situations leukocytes were shown to contribute to endothelial cell and vessel wall alterations and also to coagulation by the production of tissue factor.
This review demonstrates the important function of red blood cells in vascular disorders. Vaso-occlusion was described in sickle cell diseases as a consequence of red blood cells (RBCs) increasing their adhesion to endothelium. RBC increased adhesion was successively observed in diabetes mellitus, polycythemia vera, and retinal vascular disorders. Polycythemia vera (myeloproliferative disorder) is also complicated by a high frequency of thrombotic complications. The participation of RBCs in this process was found to be also linked to an epigenetic mutation of JAK2 kinase and to a modification of an RBC molecule involved in RBC adhesion. As for hemostasis the study of patients suffering from atherosclerotic disorders or diabetes and ageing patients allowed the discovery of new molecules of the vessel and their functions. The most recent example is the receptor for advanced glycation end products or RAGE.
2. Blood Cells and Endothelial Cells
2.1. Endothelium
Endothelial cells (ECs) are present in the inner face of the vessels. They have common characteristics but they can be specialized, in particular in the brain, kidney, liver, skin, and coronary vessels. The junctions between the endothelial cells have some differences according to their location in organs. Junctions are dynamic structures. Endothelial cells adhere to one another through junctional structures formed by transmembrane adhesive proteins. Permeability to plasma solutes is controlled, to a considerable extent, by junction permeation. Leukocyte extravasation and infiltration into inflamed areas require finely-regulated opening and closing of cell-to-cell contacts [1][2][3]. ECs participate in the regulation of the vessel tone and synthesize nitrogen oxide (NO) and prostacyclin (PGI2) [4][5]. ECs have receptors involved in blood cell–endothelial cell interactions [6]. ECs synthesize von Willebrand factor which participates in hemostasis [7].
2.2. Platelets
During the 70s, antiaggregating agents, such as aspirin, were used as a therapy in coronary disease, stroke, and peripheral vascular disease. Aspirin was shown to act by inhibiting cyclooxygenase, responsible of prostaglandin G2 (PGG2) and prostaglandin H2 (PGH2) PGG2 and PGH2 formation, which are the precursors of thromboxane A2. Anti-platelet drugs were developed and they modified platelet molecules involved in platelet activation and thrombus formation. Platelet glycoproteins (GP), GP llb and GP llla are related to membrane proteins in terms of their structure and functionality; this is also the case for their immunology. These proteins are called cytoadhesins [8] or integrins [9]. They are related to RGD-specific adhesion receptors. This also means that they have a role to play in cell-to-cell interactions. The predominant constituents of the platelet plasma membranes are formed of platelet GP IIb and IIIa (αIIbβ3). These are significant immunogens, which transport the platelet alloantigen. These are also subject to attack from allo- and autoimmune immunologic disorders. Lek (Bak) is found on GP IIb, while PLA occurs on the GP IIIa [10][11]. When we consider patients that have these platelets due to Glanzmann’s thrombasthenia (hereditary blood disorder) [12], we find that the evidence of these genetic markers is significantly limited, or possibly not present at all. This is because they lack GP IIb and IIIa. The GPIb–IX–V complex which is missing in Bernard–Soulier syndrome is the major receptor for adhesion to collagen of the sub endothelium [13]. Von Willebrand factor, when absent, is associated with a congenital hemorrhagic syndrome and is implicated in the platelet adhesion mechanism [7]. The concept that platelets may interact directly with ECs conditioned through an inflammatory environment has received increasing attention. Normally ECs present an antithrombotic surface for flowing blood by releasing nitric oxide (NO) and prostacyclin (PGI2) and through expression of CD39. Signaling into the platelet by PGI2 and NO maintains high levels of cyclic nucleotides within the platelet cytoplasm, thereby antagonizing the known pathways of platelet activation [14].
2.3. Leukocytes
The adhesion of leukocytes is able to be significantly extended when there is the presence of inflammation, infection, or heart disease [14]. The leukocyte–endothelium interaction and its molecular foundations have been the subject of considerable study in recent years [15][16][17]. Monoclonal antibodies enable us to understand the leukocyte adhesion molecule (LeuCAM). This is mediated by integrins β2 (CD18), α subunits (CD11a, CD11b, CD11c, CD11d) on leukocytes, and intercellular adhesion molecules (ICAMs) and vascular cell adhesion molecules (VCAMs) on endothelium. There is an illustration in Figure 1.
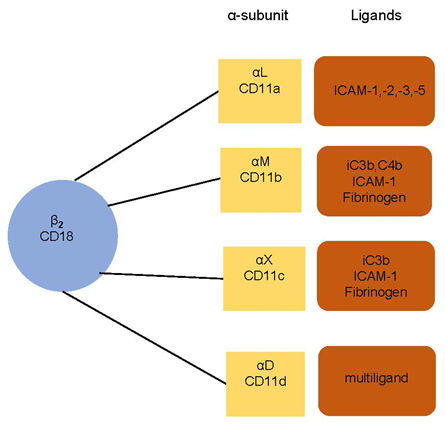
Figure 1. Leukocyte integrins and their ligands. The subunit CD18 (β2 integrins) is associated with α subunit (αL CD11a, αM CD11b, αX CD11c, and αD CD11d). The integrins bind to a specific ligand—intercellular adhesion molecules (ICAMs), fibrinogen, or complement components (iC3b, C4b, c3b). CD11d is a multiligand receptor.
The molecule interaction participates in rolling, stable adhesion, and extravasation of leukocytes. In addition to this, other leukocyte structures are connected to this process [18]. These include fibronectin receptors. Researchers have announced that there are particular structures which are connected to the endothelial cells [19]. These cells also express the intercellular adhesion molecules (ICAMs) when there is no stimulation whatsoever. However, E-selectin, and the vascular cell adhesion molecules (VCAMs) can only be located on endothelial cells which have been activated. Cytokines such as TNF-α, IL1-β, and IFN-γ, are able to induce the synthesis and expression of E-selectin and VCAMs. They can also elevate ICAMs and the human leukocyte antigens (HLAs) diabetic retinopathy (DR), DP, and DQ. Interactions between the selectin family and their respective ligands will mediate rolling adhesion of leukocytes on EC under flow conditions [14][20][21] This first step of leukocyte adhesion is then followed by interactions between members of the CD18 family with their respective ligands in the immunoglobulin family that mediate firm adhesion/arrest on leukocytes on the ECs. The leukocyte adhesion mediated by P-selectin could occur when inflammatory mediators such as histamine or thrombin activate ECs. This activation redistributes P-selectin from its storage granules to the endothelial surface. On the other hand, the expression of E-selectin on the endothelial cell surface is indeed induced only by stimulation of ECs by inflammatory cytokines (e.g., IL-1 and TNF-α). ICAMS are also major adhesion molecules that arrest circulating leukocytes on ECs.
Acute respiratory distress syndrome (ARDS), hypercholesterolemia, and diabetes mellitus are all involved in the elevation of leukocyte adhesion. This then results in vascular damage. Platelet–leukocyte aggregates occur in atherosclerosis and atherothrombotic disease. This means that researchers need to focus on modifying the leukocyte–endothelial cell in terms of anti-thrombotic and anti-atherosclerotic treatments.
Macrophage activation in atherosclerotic lesions is associated with the local release of inflammatory cytokines and reactive oxygen intermediates. During myocardial infarction, cardiomyocytes, tissue-resident macrophages, and fibroblasts passively release damage-associated molecular patterns, such as ATP and hyaluronic acid through the breakdown of the extracellular matrix. This activates local macrophages and neutrophils and stimulates a proinflammatory immune response via chemokines, cytokines, and vascular adhesion molecules through intracellular mitogen-activated protein kinases (MAPK) and NF-κB signaling [22].
SARS-CoV-2 binding to the angiotensin-converting enzyme receptor present on endothelial cells may facilitate the interaction with leukocytes, enhancing the expression of adhesion receptors. This inflammatory reaction producing interleukin-6 (IL6), macrophage chemoattractant protein-1 (MCP1), and tissue factor, can lead to thrombosis and promote apoptosis.
2.4. Red Blood Cells
2.4.1. Red Blood Cell Adhesion in Sickle Cell Anemia
Sickle cell disease (SCD) causes abnormalities in terms of function. This hematological disease showed how the RBC structure creates issues. The sickling is observed when the oxygen partial pressure is lowered, hemoglobin S polymerized, and red blood cell deformability reduced. Hebbel [23] argued that patients who suffer from sickle cell disease have issues in the vascular endothelium. An experimental model with cultured endothelial cells and red blood cells labelled with chromium-51 allows us to see the effects of this condition. During vaso-occlusive episodes these issues are elevated [24].
This sticking of RBCs to vascular walls facilitates the creation of HbS polymers. This is a result of restricting the movement of cells through the capillaries. This then has a chain reaction and causes vaso-occlusion. Many research studies have confirmed the fact that abnormal cell adhesion limits micro vessels and traps sickle cells within postcapillary venules. The movement of RBCs could be reduced in patients suffering from sickle cell disease by ensuring they remain in contact with the vascular wall by using erythroid adhesion molecules and endothelial cell proteins. Researchers have written about the interactions that occur between HbSS RBCs and the endothelial vascular wall. When α4β1 integrin (VLA-4) occurs on newly-formed moving reticulocytes, it plays a significant role as they enable RBC connection to vascular cell adhesion molecule (VCAM-1), amongst other substances [25] (see Figure 2 for more details). HbSS reticulocytes are bound to the endothelial cells using thrombospondin (TSP) which also connects their CD36 molecules [26]. That said, this does not reduce the significant impact of SCD in terms of patients who lack CD36 [27].
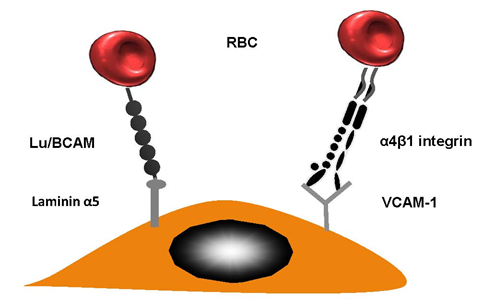
Figure 2. Red blood cell (RBC) adhesion in sickle cell diseases. Lutheran Lu/BCAM contains a particular protein which is able to forge a connection with laminin α5. The latter occurs in the sub-endothelium; however, it can also occur on the apical side endothelial cell. VLA-4 (α4β1 integrin) is a molecule produced by reticulocytes; this had a strong sticky quality.
2.4.2. Red Blood Cell Adhesion in Polycythemia Vera.
In 1892, polycythemia vera (PV) was first discovered. It was found in a person suffering from thrombosis.
It can be diagnosed through bone marrow culture, determination of red cell mass, and JAK2 mutation. The incidence of thrombotic events is very high in patients with PV. According to different studies it varies between 12% and 39% at the time of diagnosis. Recurrent thrombosis varies between 31% on the venous side and 67.6% on the arterial side. The risk of suffering from this condition is higher in patients with a high level of JAK2 V617F allele mutation [28]. JAK2 mutation occurs in more than 98% of people with PV and about 30% of patients with thrombocythemia. In patients with thrombosis of hepatic, portal, or mesenteric veins, this type of mutation was found in 34% and 45% in patients with Budd–Chiari syndrome [29].
We discovered that red blood cell (RBC) adhesion of patients with PV was increased and we explored the molecular basis of this abnormality [30] (Figure 3). Our in vitro results with PV strongly suggest that RBC–endothelium interaction may have deleterious consequences. PV RBC adhesion activates endothelial cells and stimulates vascular cell adhesion molecule expression favoring leukocyte adhesion.
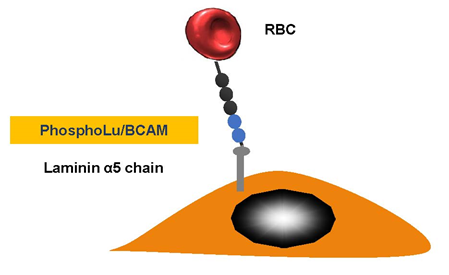
Figure 3. Polycythemia vera. Erythrocyte-phosphorylated Lu/BCAM can be found in people with polycythemia vera, as a consequence of a JAK2 V617 F mutation. This then affects the endothelial cell laminin α5, which occurs in the sub-endothelium and the apical-side endothelial cell. The Lu/BCAM cytoplasmic domain is affected by a cell adhesion to the laminin protein within a spectrin-based skeleton.
JAK2 mutations found in people suffering from PV [31][32] may create myeloproliferation. They can also cause greater numbers of blood cells and a greater likelihood of functional abnormalities. Lu/BCAM is expressed after band 3 during erythropoiesis and it first appears on early orthochromatic erythroblasts [33][34]. The fact that the disease could be easily cloned might give a reason for why its excessive adhesiveness is not limited to just reticulocytes but also involves additional moving RBCs. Patients who suffer from PV are often subject to thrombotic complications and cerebrovascular issues. Post-sinusoidal obstruction can often be found beyond the liver in the hepatic veins in patients suffering from Budd–Chiari syndrome. The variable response in flow could give an indication of why thrombosis occurs systematically.
2.4.3. Red Blood Cell Adhesion in Retinal-Vein Occlusion
Sight loss in elderly people can be caused by retinal-vein occlusion (RVO). It is the second-most-common reason for loss of sight; diabetic retinopathy is the primary cause of reduced vision. There are three sections in the anatomic classification—hemi-retinal-vein occlusion (HRVO), central-retinal-vein occlusion (CRVO), and branch-retinal-vein occlusion (BRVO). A retinal hemorrhage can indicate RVO, and so can venous dilation, which can sometimes occur with retinal swelling in a particular area. This can be diffuse for CRVO, as part of a hemiretina for HRVO or, in the case of BRVO, in connection with a retinal quadrant. BRVO is the least-serious form of RVO, which is probably due to the fact that it is found at an arteriovenous crossing [35]. The worst is central-retinal-vein occlusion (CRVO) and there is very little known about this condition. RBCs from patients with CRVO were more adherent when they express phosphatidylserine and bound to the phosphatidylserine (PS) receptor. Annexin V binding to PS RBCs inhibited adhesion to endothelial cells [36] (Figure 4). BRVO occurs at 0.6%, while CRVO is limited to 0.1%. BRVO is related to hypertension [37]. Central-retinal-artery occlusion (CRAO) is connected to eye strokes. This can appear in one in every 100,000 patients [38].
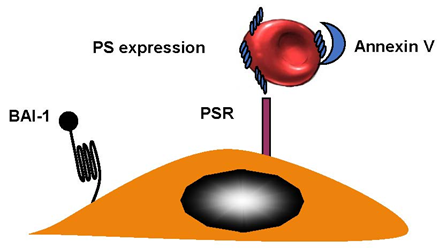
Figure 4. Retinal-vein occlusion. RBCs from patients with retinal-vein occlusion have an enhanced phosphatidylserine (PS) expression and adhere to the endothelium. Brain-specific angiogenesis inhibitor (BAI-1) and phosphatidylserine receptor (PSR), are potential receptors. Blockade of PS RBCs by annexin V or PS receptors on endothelial cells by anti PSR, inhibited adhesion indicating that the couple PS–PSR is responsible for increased adhesion of RBCs from patients with CRVO.
References
- Bazzoni, G.; Dejana, E. Endothelial cell-to-cell junctions: Molecular organization and role in vascular homeostasis. Physiol. Rev. 2004, 84, 869–901.
- DeLisser, H.M.; Newman, P.J.; Albelda, S.M. Molecular and functional aspects of PECAM-1/CD31. Immunol. Today 1994, 15, 490–495.
- Duncan, G.S.; Andrew, D.P.; Takimoto, H.; Kaufman, S.A.; Yoshida, H.; Spellberg, J. Genetic evidence for functional redundancy of Platelet/Endothelial cell adhesion molecule-1 (PECAM-1): CD31-deficient mice reveal PECAM-1-dependent and PECAM-1-independent functions. J. Immunol. 1999, 162, 3022–3030.
- Furchgott, R.F.; Zawadzki, J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980, 288, 373–376, doi:10.1038/288373a0.
- Moncada, S.; Vane, J.R. The role of prostacyclin in vascular tissue. Fed. Proc. 1979, 38, 66–71.
- Gimbrone, M.A., Jr.; Garcia-Cardena, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636, doi:10.1161/CIRCRESAHA.115.306301.
- Peyvandi, F.; Garagiola, I.; Baronciani, L. Role of von Willebrand factor in the haemostasis. Blood Transfus. 2011, 9, s3–s8, doi:10.2450/2011.002S.
- Plow, E.F.; Loftus, J.C.; Levin, E.G.; Fair, D.S.; Dixon, D.; Forsyth, J.; Ginsberg, M.H. Immunologic relationship between platelet membrane glycoprotein GPIIb/IIIa and cell surface molecules expressed by a variety of cells. Proc. Natl. Acad. Sci. USA 1986, 83, 6002–6006, doi:10.1073/pnas.83.16.6002.
- Hynes, R.O. Integrins: A family of cell surface receptors. Cell 1987, 48, 549–554, doi:10.1016/0092-8674(87)90233-9.
- Kieffer, N.; Wautier, J.L.; Coulombel, L.; Titeux, M.; Wautier, M.P.; Vainchenker, W.; Ruan, C.; Breton-Gorius, J. Uncoupling in the expression of platelet GP IIb/IIIa in human endothelial cells and K562 cells: Absence of immunologic crossreactivity between platelet GP IIb and the vitronectin receptor alpha chain. Blood 1988, 72, 1209–1215.
- Gruel, Y.; Boizard, B.; Daffos, F.; Forestier, F.; Caen, J.; Wautier, J.L. Determination of platelet antigens and glycoproteins in the human fetus. Blood 1986, 68, 488–492.
- Nurden, A.T.; Pillois, X.; Wilcox, D.A. Glanzmann thrombasthenia: State of the art and future directions. Semin. Thromb. Hemost. 2013, 39, 642–655, doi:10.1055/s-0033-1353393.
- Andrews, R.K.; Berndt, M.C. Bernard-Soulier syndrome: An update. Semin. Thromb. Hemost. 2013, 39, 656–662, doi:10.1055/s-0033-1353390.
- Ed Rainger, G.; Chimen, M.; Harrison, M.J.; Yates, C.M.; Harrison, P.; Watson, S.P.; Lordkipanidze, M.; Nash, G.B. The role of platelets in the recruitment of leukocytes during vascular disease. Platelets 2015, 26, 507–520, doi:10.3109/09537104.2015.1064881.
- Hansson, G.K.; Libby, P. The immune response in atherosclerosis: A double-edged sword. Nat. Rev. Immunol. 2006, 6, 508–519, doi:10.1038/nri1882.
- Libby, P. Mechanisms of acute coronary syndromes and their implications for therapy. N. Engl. J. Med. 2013, 368, 2004–2013, doi:10.1056/NEJMra1216063.
- Wautier, J.L.; Setiadi, H.; Vilette, D.; Weill, D.; Wautier, M.P. Leukocyte adhesion to endothelial cells. Biorheology 1990, 27, 425–432, doi:10.3233/bir-1990-273-419.
- Barbe, L.L.; Boval, B.M.; Wautier, M.P.; Wautier, J.L. An in vitro system for testing leucocyte and leukaemic cell line adhesion to synthetic fibres. Br. J. Haematol. 2001, 115, 664–671, doi:10.1046/j.1365-2141.2001.03137.x.
- Liote, F.; Boval-Boizard, B.; Weill, D.; Kuntz, D.; Wautier, J.L. Blood monocyte activation in rheumatoid arthritis: Increased monocyte adhesiveness, integrin expression, and cytokine release. Clin. Exp. Immunol. 1996, 106, 13–19, doi:10.1046/j.1365-2249.1996.d01-820.x.
- Nash, G.B. Adhesion between platelets and leukocytes or endothelial cells. Methods Mol. Biol. 2004, 272, 199–213, doi:10.1385/1-59259-782-3:199.
- Setiadi, H.; McEver, R.P. Clustering endothelial E-selectin in clathrin-coated pits and lipid rafts enhances leukocyte adhesion under flow. Blood 2008, 111, 1989–1998, doi:10.1182/blood-2007-09-113423.
- Jones, D.P.; True, H.D.; Patel, J. Leukocyte Trafficking in Cardiovascular Disease: Insights from Experimental Models. Mediat. Inflamm. 2017, 2017, 9746169, doi:10.1155/2017/9746169.
- Hebbel, R.P.; Boogaerts, M.A.; Eaton, J.W.; Steinberg, M.H. Erythrocyte adherence to endothelium in sickle-cell anemia. A possible determinant of disease severity. N. Engl. J. Med. 1980, 302, 992–995, doi:10.1056/NEJM198005013021803.
- Wautier, J.L.; Galacteros, F.; Wautier, M.P.; Pintigny, D.; Beuzard, Y.; Rosa, J.; Caen, J.P. Clinical manifestations and erythrocyte adhesion to endothelium in sickle cell syndrome. Am. J. Hematol. 1985, 19, 121–130, doi:10.1002/ajh.2830190203.
- El Nemer, W.; Wautier, M.P.; Rahuel, C.; Gane, P.; Hermand, P.; Galacteros, F.; Wautier, J.L.; Cartron, J.P.; Colin, Y.; Le Van Kim, C. Endothelial Lu/BCAM glycoproteins are novel ligands for red blood cell alpha4beta1 integrin: Role in adhesion of sickle red blood cells to endothelial cells. Blood 2007, 109, 3544–3551, doi:10.1182/blood-2006-07-035139.
- Sugihara, K.; Sugihara, T.; Mohandas, N.; Hebbel, R.P. Thrombospondin mediates adherence of CD36+ sickle reticulocytes to endothelial cells. Blood 1992, 80, 2634–2642.
- Lee, K.; Gane, P.; Roudot-Thoraval, F.; Godeau, B.; Bachir, D.; Bernaudin, F.; Cartron, J.P.; Galacteros, F.; Bierling, P. The nonexpression of CD36 on reticulocytes and mature red blood cells does not modify the clinical course of patients with sickle cell anemia. Blood 2001, 98, 966–971, doi:10.1182/blood.v98.4.966.
- Vannucchi, A.M.; Antonioli, E.; Guglielmelli, P.; Longo, G.; Pancrazzi, A.; Ponziani, V.; Bogani, C.; Ferrini, P.R.; Rambaldi, A.; Guerini, V.; et al. Prospective identification of high-risk polycythemia vera patients based on JAK2(V617F) allele burden. Leukemia 2007, 21, 1952–1959, doi:10.1038/sj.leu.2404854.
- Kiladjian, J.J.; Cervantes, F.; Leebeek, F.W.; Marzac, C.; Cassinat, B.; Chevret, S.; Cazals-Hatem, D.; Plessier, A.; Garcia-Pagan, J.C.; Darwish Murad, S.; et al. The impact of JAK2 and MPL mutations on diagnosis and prognosis of splanchnic vein thrombosis: A report on 241 cases. Blood 2008, 111, 4922–4929, doi:10.1182/blood-2007-11-125328.
- Wautier, M.P.; El Nemer, W.; Gane, P.; Rain, J.D.; Cartron, J.P.; Colin, Y.; Le Van Kim, C.; Wautier, J.L. Increased adhesion to endothelial cells of erythrocytes from patients with polycythemia vera is mediated by laminin alpha5 chain and Lu/BCAM. Blood 2007, 110, 894–901, doi:10.1182/blood-2006-10-048298.
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061, doi:10.1016/S0140-6736(05)71142-9.
- James, C.; Ugo, V.; Le Couedic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garcon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148, doi:10.1038/nature03546.
- Bony, V.; Gane, P.; Bailly, P.; Cartron, J.P. Time-course expression of polypeptides carrying blood group antigens during human erythroid differentiation. Br. J. Haematol. 1999, 107, 263–274, doi:10.1046/j.1365-2141.1999.01721.x.
- Southcott, M.J.; Tanner, M.J.; Anstee, D.J. The expression of human blood group antigens during erythropoiesis in a cell culture system. Blood 1999, 93, 4425–4435.
- Nentwich, M.M.; Ulbig, M.W. Diabetic retinopathy—ocular complications of diabetes mellitus. World J Diabetes 2015, 6, 489–499, doi:10.4239/wjd.v6.i3.489.
- Wautier, M.P.; Heron, E.; Picot, J.; Colin, Y.; Hermine, O.; Wautier, J.L. Red blood cell phosphatidylserine exposure is responsible for increased erythrocyte adhesion to endothelium in central retinal vein occlusion. J. Thromb. Haemost. 2011, 9, 1049–1055, doi:10.1111/j.1538-7836.2011.04251.x.
- Klein, R.; Klein, B.E.; Moss, S.E.; Meuer, S.M. The epidemiology of retinal vein occlusion: The Beaver Dam Eye Study. Trans. Am. Ophthalmol. Soc. 2000, 98, 133–141; discussion 141-133.
- Varma, D.D.; Cugati, S.; Lee, A.W.; Chen, C.S. A review of central retinal artery occlusion: Clinical presentation and management. Eye (Lond) 2013, 27, 688–697, doi:10.1038/eye.2013.25.