A basic requirement of tumorigenesis is the development of a vascular network to support the metabolic requirements of tumor growth and metastasis. Tumor vascular formation is regulated by a balance between promoters and inhibitors of angiogenesis. Typically, the pro-angiogenic environment created by the tumor is extremely aggressive, resulting in the rapid vessel formation with abnormal, dysfunctional morphology. The altered morphology and function of tumor blood and lymphatic vessels has numerous implications including poor perfusion, tissue hypoxia, and reduced therapy uptake. Targeting tumor angiogenesis as a therapeutic approach has been pursued in a host of different cancers. Although some preclinical success was seen, there has been a general lack of clinical success with traditional anti-angiogenic therapeutics as single agents. Typically, following anti-angiogenic therapy, there is remodeling of the tumor microenvironment and widespread tumor hypoxia, which is associated with development of therapy resistance. A more comprehensive understanding of the biology of tumor angiogenesis and insights into new clinical approaches, including combinations with immunotherapy, are needed to advance vascular targeting as a therapeutic area.
- hypoxia
- angiogenesis
- vascular normalization
1. Introduction
1.1. Sprouting Angiogenesis in Normal Physiology
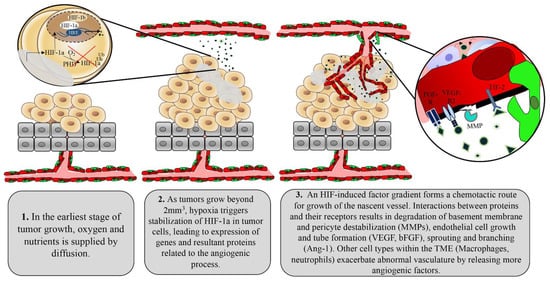
1.2. Tumor Control of Angiogenesis
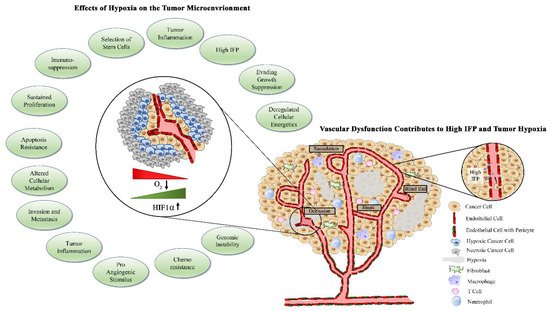
1.3. Factors Contributing to Tumor Vascular Dysfunction
1.4. Abnormal Vasculature Results in Limited Treatment Delivery
2. Vascular Normalizing Agents as Adjuvants to Traditional Cancer Therapeutics
References
- Pereira, R.D.; de Long, N.E.; Wang, R.C.; Yazdi, F.T.; Holloway, A.C.; Raha, S. Angiogenesis in the Placenta: The Role of Reactive Oxygen Species Signaling. BioMed Res. Int. 2015, 2015, 814543.
- Dangat, K.; Khaire, A.; Joshi, S. Cross Talk of Vascular Endothelial Growth Factor and Neurotrophins in Mammary Gland Development. Growth Factors 2020, 38, 16–24.
- Kumar, P.; Kumar, S.; Udupa, E.P.; Kumar, U.; Rao, P.; Honnegowda, T. Role of Angiogenesis and Angiogenic Factors in Acute and Chronic Wound Healing. Plast. Aesthet. Res. 2015, 2, 243–249.
- Ucuzian, A.A.; Gassman, A.A.; East, A.T.; Greisler, H.P. Molecular Mediators of Angiogenesis. J. Burn Care Res. 2010, 31, 158–175.
- Yokota, Y.; Nakajima, H.; Wakayama, Y.; Muto, A.; Kawakami, K.; Fukuhara, S.; Mochizuki, N. Endothelial Ca2+ Oscillations Reflect VEGFR Signaling-Regulated Angiogenic Capacity in Vivo. eLife 2015, 4, e08817.
- Ghajar, C.M.; George, S.C.; Putnam, A.J. Matrix Metalloproteinase Control of Capillary Morphogenesis. Crit. Rev. Eukaryot. Gene Expr. 2008, 18, 251–278.
- Sauteur, L.; Krudewig, A.; Herwig, L.; Ehrenfeuchter, N.; Lenard, A.; Affolter, M.; Belting, H.G. Cdh5/VE-Cadherin Promotes Endothelial Cell Interface Elongation via Cortical Actin Polymerization during Angiogenic Sprouting. Cell Rep. 2014, 9, 504–513.
- Franco, C.A.; Jones, M.L.; Bernabeu, M.O.; Vion, A.C.; Barbacena, P.; Fan, J.; Mathivet, T.; Fonseca, C.G.; Ragab, A.; Yamaguchi, T.P.; et al. Non-Canonical Wnt Signalling Modulates the Endothelial Shear Stress Flow Sensor in Vascular Remodelling. eLife 2016, 5, e07727.
- Chen, Q.; Jiang, L.; Li, C.; Hu, D.; Bu, J.W.; Cai, D.; Du, J.L. Haemodynamics-Driven Developmental Pruning of Brain Vasculature in Zebrafish. PLoS Biol. 2012, 10, e1001374.
- Murakami, M. Signaling Required for Blood Vessel Maintenance: Molecular Basis and Pathological Manifestations. Int. J. Vasc. Med. 2012, 2012, 293641.
- Bremnes, R.M.; Dønnem, T.; Al-Saad, S.; Al-Shibli, K.; Andersen, S.; Sirera, R.; Camps, C.; Marinez, I.; Busund, L.T. The Role of Tumor Stroma in Cancer Progression and Prognosis: Emphasis on Carcinoma-Associated Fibroblasts and Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2011, 6, 209–217.
- Murphy, K.J.; Chambers, C.R.; Herrmann, D.; Timpson, P.; Pereira, B.A. Dynamic Stromal Alterations Influence Tumor-Stroma Crosstalk to Promote Pancreatic Cancer and Treatment Resistance. Cancers 2021, 13, 3481.
- Folkman, J. Tumor Angiogenesis: Therapeutic Implications. N. Engl. J. Med. 1971, 285, 1182–1186.
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70.
- Folkman, J.; Hanahan, D. Switch to the Angiogenic Phenotype during Tumorigenesis. Princess Takamatsu Symp. 1991, 22, 22.
- Comerford, K.M.; Wallace, T.J.; Karhausen, J.; Louis, N.A.; Montalto, M.C.; Colgan, S.P. Hypoxia-Inducible Factor-1-Dependent Regulation of the Multidrug Resistance (MDR1) Gene. Cancer Res. 2002, 62, 62.
- Wilkins, S.E.; Abboud, M.I.; Hancock, R.L.; Schofield, C.J. Targeting Protein-Protein Interactions in the HIF System. ChemMedChem 2016, 11, 773–786.
- Jeong, J.-W.; Bae, M.-K.; Ahn, M.-Y.; Kim, S.-H.; Sohn, T.-K.; Bae, M.-H.; Yoo, M.-A.; Song, E.J.; Lee, K.-J.; Kim, K.-W. Regulation and Destabilization of HIF-1 by ARD1-Mediated Acetylation Quitin-Proteasome Pathway (Salceda and Caro The Association of PVHL and HIF-1 under nor-Moxic Conditions Is Triggered by the Posttranslational. Cell 2002, 111, 709–720.
- Strowitzki, M.; Cummins, E.; Taylor, C. Protein Hydroxylation by Hypoxia-Inducible Factor (HIF) Hydroxylases: Unique or Ubiquitous? Cells 2019, 8, 384.
- Yu, A.Y.; Frid, M.G.; Shimoda, L.A.; Wiener, C.M.; Stenmark, K.; Semenza, G.L. Temporal, Spatial, and Oxygen-Regulated Expression of Hypoxia-Inducible Factor-1 in the Lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 1998, 275, L818–L826.
- McKeown, S.R. Defining Normoxia, Physoxia and Hypoxia in Tumours—Implications for Treatment Response. Br. J. Radiol. 2014, 87, 20130676.
- Artemov, A.V.; Zhigalova, N.; Zhenilo, S.; Mazur, A.M.; Prokhortchouk, E.B. VHL Inactivation without Hypoxia Is Sufficient to Achieve Genome Hypermethylation. Sci. Rep. 2018, 8, 10667.
- Amelio, I.; Mancini, M.; Petrova, V.; Cairns, R.A.; Vikhreva, P.; Nicolai, S.; Marini, A.; Antonov, A.A.; le Quesne, J.; Baena Acevedo, J.D.; et al. P53 Mutants Cooperate with HIF-1 in Transcriptional Regulation of Extracellular Matrix Components to Promote Tumor Progression. Proc. Natl. Acad. Sci. USA 2018, 115, E10869–E10878.
- Ravi, R.; Mookerjee, B.; Bhujwalla, Z.M.; Sutter, C.H.; Artemov, D.; Zeng, Q.; Dillehay, L.E.; Madan, A.; Semenza, G.L.; Bedi, A. Regulation of Tumor Angiogenesis by P53-Induced Degradation of Hypoxia- Inducible Factor 1α. Genes Dev. 2000, 14, 34–44.
- Mohlin, S.; Hamidian, A.; von Stedingk, K.; Bridges, E.; Wigerup, C.; Bexell, D.; Påhlman, S. PI3K-MTORC2 but Not PI3K-MTORC1 Regulates Transcription of HIF2A/EPAS1and Vascularization in Neuroblastoma. Cancer Res. 2015, 75, 4617–4628.
- Zhong, H.; Chiles, K.; Feldser, D.; Laughner, E.; Hanrahan, C.; Georgescu, M.M.; Simons, J.W.; Semenza, G.L. Modulation of Hypoxia-Inducible Factor 1α Expression by the Epidermal Growth Factor/Phosphatidylinositol 3-Kinase/PTEN/AKT/FRAP Pathway in Human Prostate Cancer Cells: Implications for Tumor Angiogenesis and Therapeutics. Cancer Res. 2000, 60, 1541–1545.
- Lee, B.I.; Kim, W.H.; Jung, J.; Cho, S.J.; Park, J.W.; Kim, J.; Chung, H.Y.; Chang, M.S.; Nam, S.Y. A Hypoxia-Independent up-Regulation of Hypoxia-Inducible Factor-1 by AKT Contributes to Angiogenesis in Human Gastric Cancer. Carcinogenesis 2008, 29, 44–51.
- Stegeman, H.; Span, P.N.; Peeters, W.J.M.; Verheijen, M.M.G.; Grénman, R.; Meijer, T.W.H.; Kaanders, J.H.A.M.; Bussink, J. Interaction between Hypoxia, AKT and HIF-1 Signaling in HNSCC and NSCLC: Implications for Future Treatment Strategies. Future Sci. OA 2016, 2.
- Ferrara, N. Vascular Endothelial Growth Factor: Basic Science and Clinical Progress. Endocr. Rev. 2004, 25, 581–611.
- Claesson-Welsh, L.; Welsh, M. VEGFA and Tumour Angiogenesis. J. Intern. Med. 2013, 273, 114–127.
- Li, X.; Eriksson, U. Novel VEGF Family Members: VEGF-B, VEGF-C and VEGF-D. Int. J. Biochem. Cell Biol. 2001, 33, 421–426.
- Liang, L.; Yue, Z.; Du, W.; Li, Y.; Tao, H.; Wang, D.; Wang, R.; Huang, Z.; He, N.; Xie, X.; et al. Molecular Imaging of Inducible VEGF Expression and Tumor Progression in a Breast Cancer Model. Cell. Physiol. Biochem. 2017, 42, 407–415.
- Chang, J.; Chaudhuri, O. Beyond Proteases: Basement Membrane Mechanics and Cancer Invasion. J. Cell Biol. 2019, 218, 2456–2469.
- Zucker, S.; Vacirca, J. Role of Matrix Metalloproteinases (MMPs) in Colorectal Cancer. Cancer Metastasis Rev. 2004, 23, 101–117.
- Hashizume, H.; Baluk, P.; Morikawa, S.; McLean, J.W.; Thurston, G.; Roberge, S.; Jain, R.K.; McDonald, D.M. Openings between Defective Endothelial Cells Explain Tumor Vessel Leakiness. Am. J. Pathol. 2000, 156, 1363–1380.
- Hida, K.; Hida, Y.; Amin, D.N.; Flint, A.F.; Panigrahy, D.; Morton, C.C.; Klagsbrun, M. Tumor-Associated Endothelial Cells with Cytogenetic Abnormalities. Cancer Res. 2004, 64, 8249–8255.
- Schaaf, M.B.; Garg, A.D.; Agostinis, P. Defining the Role of the Tumor Vasculature in Antitumor Immunity and Immunotherapy Article. Cell Death Dis. 2018, 9, 115.
- Matsuda, K.; Ohga, N.; Hida, Y.; Muraki, C.; Tsuchiya, K.; Kurosu, T.; Akino, T.; Shih, S.C.; Totsuka, Y.; Klagsbrun, M.; et al. Isolated Tumor Endothelial Cells Maintain Specific Character during Long-Term Culture. Biochem. Biophys. Res. Commun. 2010, 394, 947–954.
- Sievert, W.; Tapio, S.; Breuninger, S.; Gaipl, U.; Andratschke, N.; Trott, K.R.; Multhoff, G. Adhesion Molecule Expression and Function of Primary Endothelial Cells in Benign and Malignant Tissues Correlates with Proliferation. PLoS ONE 2014, 9, e91808.
- Ohmura-Kakutani, H.; Akiyama, K.; Maishi, N.; Ohga, N.; Hida, Y.; Kawamoto, T.; Iida, J.; Shindoh, M.; Tsuchiya, K.; Shinohara, N.; et al. Identification of Tumor Endothelial Cells with High Aldehyde Dehydrogenase Activity and a Highly Angiogenic Phenotype. PLoS ONE 2014, 9, e113910.
- Mehran, R.; Nilsson, M.; Khajavi, M.; Du, Z.; Cascone, T.; Wu, H.K.; Cortes, A.; Xu, L.; Zurita, A.; Schier, R.; et al. Tumor Endothelial Markers Define Novel Subsets of Cancer-Specific Circulating Endothelial Cells Associated with Antitumor Efficacy. Cancer Res. 2014, 74, 2731–2741.
- Gatmaitan, Z.; Varticovski, L.; Ling, L.; Mikkelsen, R.; Steffan, A.M.; Arias, I.M. Studies on Fenestral Contraction in Rat Liver Endothelial Cells in Culture. Am. J. Pathol. 1996, 148, 2027–2041.
- Levick, J.R.; Smaje, L.H. An Analysis of the Permeability of a Fenestra. Microvasc. Res. 1987, 33, 233–256.
- Suarez, S.; Ballmer-Hofer, K. VEGF Transiently Disrupts Gap Junctional Communication in Endothelial Cells. J. Cell Sci. 2001, 114, 1229–1235.
- Nimlamool, W.; Andrews, R.M.K.; Falk, M.M. Connexin43 Phosphorylation by PKC and MAPK Signals VEGF-Mediated Gap Junction Internalization. Mol. Biol. Cell 2015, 26, 2755–2768.
- Thuringer, D. The Vascular Endothelial Growth Factor-Induced Disruption of Gap Junctions is Relayed by an Autocrine Communication via ATP Release in Coronary Capillary Endothelium. Ann. N. Y. Acad. Sci. 2004, 1030, 14–27.
- Dvorak, H.F.; Senger, D.R.; Dvorak, A.M. Fibrin as a Component of the Tumor Stroma: Origins and Biological Significance. Cancer Metastasis Rev. 1983, 2, 41–73.
- Xiang, D.; Feng, Y.; Wang, J.; Zhang, X.; Shen, J.; Zou, R.; Yuan, Y. Platelet-derived Growth Factor-BB Promotes Proliferation and Migration of Retinal Microvascular Pericytes by Up-regulating the Expression of C-X-C Chemokine Receptor Types 4. Exp. Ther. Med. 2019, 18, 4022–4030.
- Lindahl, P.; Johansson, B.R.; Levéen, P.; Betsholtz, C. Pericyte Loss and Microaneurysm Formation in PDGF-B-Deficient Mice. Science 1997, 277, 242–245.
- Franco, M.; Roswall, P.; Cortez, E.; Hanahan, D.; Pietras, K. Pericytes Promote Endothelial Cell Survival through Induction of Autocrine VEGF-Asignaling and Bcl-w Expression. Blood 2011, 118, 2906–2917.
- Díaz-Flores, L.; Gutiérrez, R.; Madrid, J.F.; Varela, H.; Valladares, F.; Acosta, E.; Martín-Vasallo, P.; Díaz-Flores, J. Pericytes. Morphofunction, Interactions and Pathology in a Quiescent and Activated Mesenchymal Cell Niche. Histol. Histopathol. 2009, 24, 909–969.
- Jain, R.K. Transport of Molecules in the Tumor Interstitium: A Review. Cancer Res. 1987, 47, 3039–3051.
- Libutti, S.K.; Tamarkin, L.; Nilubol, N. Targeting the Invincible Barrier for Drug Delivery in Solid Cancers: Interstitial Fluid Pressure. Oncotarget 2018, 9, 35723–35725.
- Griffon-Etienne, G.; Boucher, Y.; Brekken, C.; Suit, H.D.; Jain, R.K. Taxane-Induced Apoptosis Decompresses Blood Vessels and Lowers Interstitial Fluid Pressure in Solid Tumors: Clinical Implications. Cancer Res. 1999, 59, 59.
- Padera, T.P.; Stoll, B.R.; Tooredman, J.B.; Capen, D.; di Tomaso, E.; Jain, R.K. Pathology: Cancer Cells Compress Intratumour Vessels 1 11672. Nature 2004, 427, 695.
- Weis, S.M.; Cheresh, D.A. Pathophysiological Consequences of VEGF-Induced Vascular Permeability. Nature 2005, 437, 497–504.
- Nagy, J.A.; Dvorak, A.M.; Dvorak, H.F. Vascular Hyperpermeability, Angiogenesis, and Stroma Generation. Cold Spring Harb. Perspect. Med. 2012, 2, a006544.
- Stylianopoulos, T.; Martin, J.D.; Chauhan, V.P.; Jain, S.R.; Diop-Frimpong, B.; Bardeesy, N.; Smith, B.L.; Ferrone, C.R.; Hornicek, F.J.; Boucher, Y.; et al. Causes, Consequences, and Remedies for Growth-Induced Solid Stress in Murine and Human Tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 15101–15108.
- Helmlinger, G.; Yuan, F.; Dellian, M.; Jain, R.K. Interstitial PH and PO2 Gradients in Solid Tumors in Vivo: High-Resolution Measurements Reveal a Lack of Correlation. Nat. Med. 1997, 3, 177–182.
- Simonsen, T.G.; Lund, K.V.; Hompland, T.; Kristensen, G.B.; Rofstad, E.K. DCE-MRI–Derived Measures of Tumor Hypoxia and Interstitial Fluid Pressure Predict Outcomes in Cervical Carcinoma. Int. J. Radiat. Oncol. Biol. Phys. 2018, 102, 1193–1201.
- Ferretti, S.; Allegrini, P.R.; Becquet, M.M.; McSheehy, P.M.J. Tumor Interstitial Fluid Pressure as an Early-Response Marker for Anticancer Therapeutics. Neoplasia 2009, 11, 874–881.
- Baish, J.W.; Netti, P.A.; Jain, R.K. Transmural Coupling of Fluid Flow in Microcirculatory Network and Interstitium in Tumors. Microvasc. Res. 1997, 53, 128–141.
- Wu, M.; Frieboes, H.B.; Chaplain, M.A.J.; McDougall, S.R.; Cristini, V.; Lowengrub, J.S. The Effect of Interstitial Pressure on Therapeutic Agent Transport: Coupling with the Tumor Blood and Lymphatic Vascular Systems. J. Theor. Biol. 2014, 355, 194–207.
- Jain, R.K.; Stylianopoulos, T. Delivering Nanomedicine to Solid Tumors. Nat. Rev. Clin. Oncol. 2010, 7, 653–664.
- Bender, L.H.; Abbate, F.; Walters, I.B. Intratumoral Administration of a Novel Cytotoxic Formulation with Strong Tissue Dispersive Properties Regresses Tumor Growth and Elicits Systemic Adaptive Immunity in in Vivo Models. Int. J. Mol. Sci. 2020, 21, 4493.
- Sriraman, S.K.; Aryasomayajula, B.; Torchilin, V.P. Barriers to Drug Delivery in Solid Tumors. Tissue Barriers 2014, 2, e29528.
- Mpekris, F.; Voutouri, C.; Baish, J.W.; Duda, D.G.; Munn, L.L.; Stylianopoulos, T.; Jain, R.K. Combining Microenvironment Normalization Strategies to Improve Cancer Immunotherapy. Proc. Natl. Acad. Sci. USA 2020, 117, 3728–3737.
- Kłosowska-Wardȩga, A.; Hasumi, Y.; Burmakin, M.; Åhgren, A.; Stuhr, L.; Moen, I.; Reed, R.K.; Rubin, K.; Hellberg, C.; Heldin, C.H. Combined Anti-Angiogenic Therapy Targeting PDGF and Vegf Receptors Lowers the Interstitial Fluid Pressure in a Murine Experimental Carcinoma. PLoS ONE 2009, 4, e8149.
- Chouaib, S.; Noman, M.Z.; Kosmatopoulos, K.; Curran, M.A. Hypoxic Stress: Obstacles and Opportunities for Innovative Immunotherapy of Cancer. Oncogene 2017, 36, 439–445.
- Matuszewska, K.; Santry, L.A.; van Vloten, J.P.; AuYeung, A.W.K.; Major, P.P.; Lawler, J.; Wootton, S.K.; Bridle, B.W.; Petrik, J. Combining Vascular Normalization with an Oncolytic Virus Enhances Immunotherapy in a Preclinical Model of Advanced-Stage Ovarian Cancer. Clin. Cancer Res. 2019, 25, 1624–1638.
- Shrimali, R.K.; Yu, Z.; Theoret, M.R.; Chinnasamy, D.; Restifo, N.P.; Rosenberg, S.A. Antiangiogenic Agents Can Increase Lymphocyte Infiltration into Tumor and Enhance the Effectiveness of Adoptive Immunotherapy of Cancer. Cancer Res. 2010, 70, 6171–6180.
- Tian, L.; Goldstein, A.; Wang, H.; Lo, H.C.; Kim, I.S.; Welte, T.; Sheng, K.; Dobrolecki, L.E.; Zhang, X.; Putluri, N.; et al. Mutual Regulation of Tumour Vessel Normalization and Immunostimulatory Reprogramming. Nature 2017, 544, 250–254.
- Goel, S.; Wong, A.H.K.; Jain, R.K. Vascular Normalization as a Therapeutic Strategy for Malignant and Nonmalignant Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006486.