In recent years, the incidence of immune-mediated gastrointestinal disorders, including celiac disease (CeD) and inflammatory bowel disease (IBD), is increasingly growing worldwide. It is well established that primary immunodeficiencies (PIDs) exhibit gastrointestinal manifestations and mimic other diseases, including CeD and IBD. The most common PIDs in adults are the selective immunoglobulin A deficiency (SIgAD) and the common variable immunodeficiency (CVID). However, some differences concerning diagnostics and management between enteropathy/colitis in PIDs, as compared to idiopathic forms of CeD/IBD, have been described. There is an ongoing discussion whether CeD and IBD in CVID patients should be considered a true CeD and IBD or just CeD-like and IBD-like diseases. This review addresses the current state of the art of the most common primary immunodeficiencies in adults and co-occurring CeD and IBD.
- primary immunodeficiency
- selective IgA deficiency
- common variable immunodeficiency
- celiac disease
- inflammatory bowel disease
- Crohn’s disease
- ulcerative colitis
1. Introduction
Autoimmune diseases of the gastrointestinal (GI) tract are increasingly growing worldwide over the last decades. They concern both inflammatory bowel disease (IBD) and celiac disease (CeD) [1][2]. The increasing body of evidence that primary immunodeficiency (PID) can complicate diagnostics of CeD [3] and mimic IBD [4] implicates the need for a comprehensive review of this topic, especially when several studies have shown that autoimmune manifestations are the second most common manifestation of PIDs after infections [5][6].
PIDs are often considered pediatric ailments, whereas between 25 and 45% of PIDs are diagnosed in adults. Among all PIDs, more than 50% make up abnormalities in humoral immunity [7], making immunoglobulin deficiency the most common PID in children and also among adults. In the latter group, selective immunoglobulin A deficiency (SIgAD) and common variable immunodeficiency (CVID) are the most common diagnoses [8].
Here we aim to present the up-to-date knowledge on the incidence, pathophysiology, symptoms, diagnostics, and management of autoimmune GI diseases, specifically CeD and IBD, in patients with underlying SIgAD or CVID. Moreover, this review is focused on differences between the classic forms of the above-mentioned diseases and those observed in patients with compromised humoral immunity, to estimate whether we are facing a spectrum of one disease or different diseases characterized by a similar clinical manifestation.
2. Immune-mediated gastrointestinal diseases in adults with primary humoral immunodeficiencies
2.1. Celiac Disease in Selective IgA Deficiency
Various sources seem to agree on the occurrence of SIgAD among CeD patients, which is approximately 2–3%. Such a value is provided by the ESsCD guidelines for 2020 and is based on a large European cohort study by McGowan et al. [9][10]. The prevalence of CeD in individuals with SIgAD varies depending on author (6.7-30%) [10][11][12][13][14]. Most authors agree that the prevalence of CeD in patients with SIgAD is higher than in the general population; however, this requires further investigation.
The basic principles of CeD diagnostics in SIgAD patients are similar to those applied to IgA-sufficient individuals. The most important difference concerns serological diagnostics. In immunocompetent patients, first-line testing comprises of measurement of the anti-TG2 IgA level in serum [9]. This approach is not effective in SIgAD patients, as false negative results might be obtained; thus, patients with SIgAD may elude CeD diagnosis [15][16][17]. To avoid such a situation, ESsCD recommended assessing total IgA levels concurrently with serology testing to determine whether IgA levels are sufficient [9]. In the case of a lowered IgA serum level, IgG anti-TG2 and IgG-DGP are regarded as the best tool for identifying CeD in patients [9][18]. The next step in CeD diagnostics, after serological examination, is a small intestinal biopsy and histological assessment of the taken specimens. Histopathology findings in SIgAD patients with CeD do not vary much from those of patients with CeD alone [19][20]. It is indistinguishable from the pathology seen in non-SIgAD patients with CeD in terms of increased numbers of intraepithelial lymphocytes, villous atrophy, crypt hyperplasia, and infiltration of the lamina propria with lymphoid cells. The main difference concerns IgA-secreting plasma cells, which are absent in CeD patients with coexisting SIgAD [21][22][23]. Symptoms of CeD in IgA-deficient patients do not vary from those in patients with CeD alone [4]. SIgAD patients with CeD are responsive to gluten withdrawal and a gluten-free diet (GFD) is recommended [24][25][26][27].
2.2. Celiac Disease in Common Variable Immunodeficiency
The prevalence of enteropathy in CVID patients varies depending on the author (10 up to 60%) [28][29][30]. These values concern the incidence of non-infectious enteropathy, which is not synonymous with CeD. The true prevalence of the latter is very difficult to estimate, as it is not fully established whether we are facing CeD or only celiac-like enteropathy in CVID patients. Regardless of the way we classify this entity, CeD-resembling findings, including villus atrophy and increased intraepithelial lymphocytes, are common among CVID patients (8-50%) [31][32][33][34][35].
Enteropathy accompanied by malabsorption is a common clinical presentation of CVID; however, villus atrophy remains a challenging finding in this group. Serological diagnostics is insufficient in individuals with CVID [26][36][26]. In the vast majority of cases, CVID enteropathy differs from CeD not only the results of the serological assays, so it is debated whether it can be classified as true CeD or rather should be called celiac like [32]. Histological assessment of a duodenal biopsy shows a paucity or absence of plasma cells [25]. Besides, villous atrophy is usually less severe and the IEL count is often lower than in CeD, excessive neutrophil infiltration can be observed, as well as graft versus host-like lesions or follicular lymphoid hyperplasia [28][32]. ESsCD guidelines from 2019 say directly that an absence of plasma cells suggests CVID [9]. It is suggested that the only criterion to confirm CeD diagnosis in CVID patients is the histological response to a GFD [37][38]. Other researchers highlighted the importance of HLA determination, as the presence of the DQ2 or DQ8 haplotype can be associated with concomitant CeD, thus allowing the identification of CVID patients at risk [35]. Furthermore, if both HLA-DQ2 and HLA-DQ8 are negative, the diagnosis of CeD is highly unlikely [9]. There is no doubt that true CeD can be diagnosed in CVID patients, but yet according to current knowledge, this relationship is rare. In the vast literature research, we found no data on differences between symptoms of CVID enteropathy and symptoms of CeD, which led us to the conclusion that manifestations of these entities are indistinguishable based on clinical signs.
Approximately 50% of patients do not respond to GFD [39]. Some sources indicate that this percentage might be even up to 80% [28]. GFD is recommended only in HLA-DQ2 or HLA-DQ8 carriers as an attempt of a 6- to 12-month trial with follow-up histologic assessment [6][28][32]. Patients without HLA-DQ2 or HLA-DQ8 or unresponsive to GFD should be treated as a distinct disorder. In this case, short-term steroids or immunomodulators, including azathioprine and 6-mercaptopurine, can be used [25]. There is also evidence for the efficacy of an elemental diet, while in the case of severe malabsorption, limited use of total parenteral nutrition might be required [11][25].
A graphical summary concerning CeD in the discussed PIDs is presented in Figure 1.
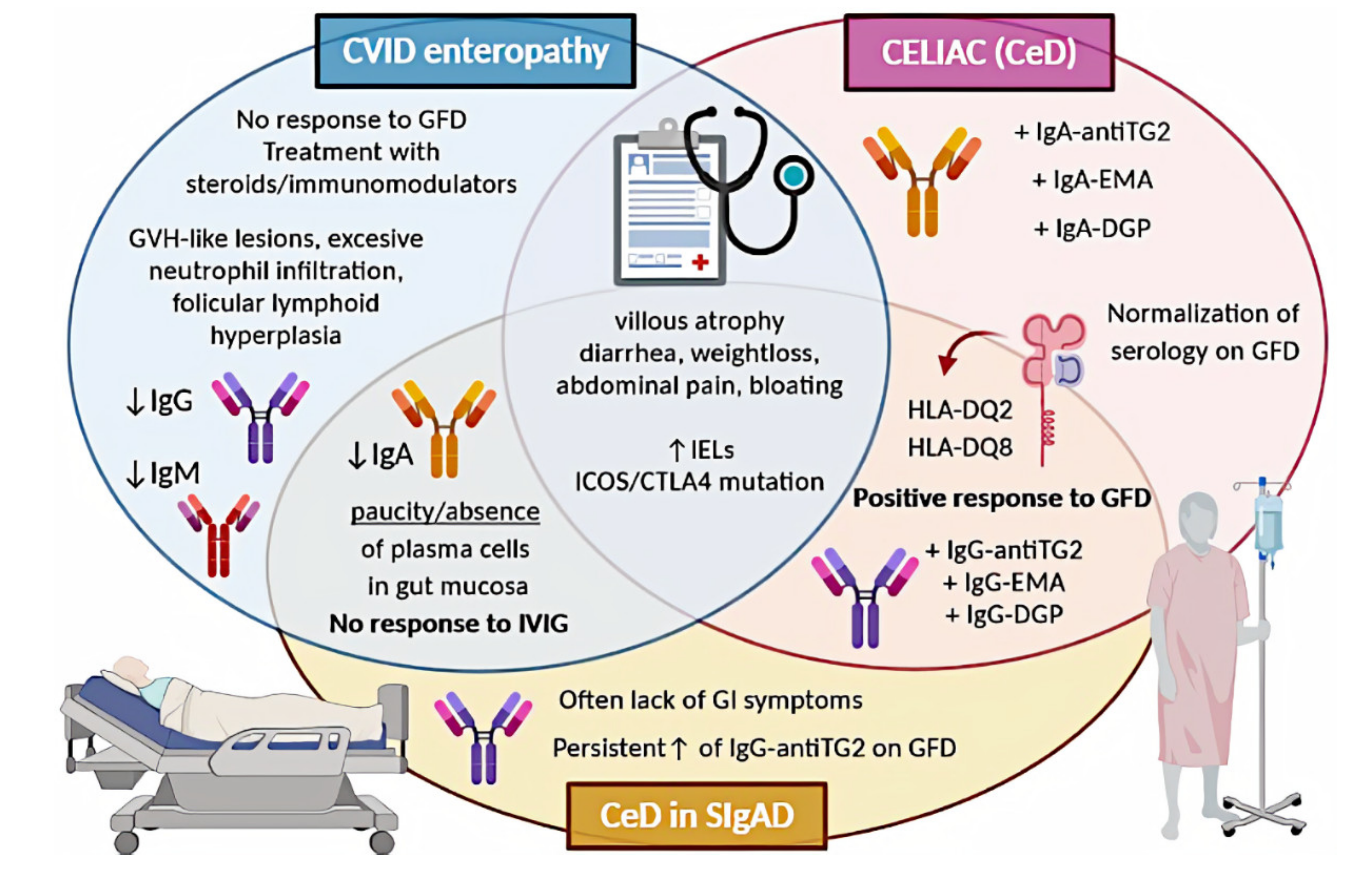
Figure 1. Comparison of CeD, CeD in SIgAD, and CVID enteropathy. CVID, common variable immunodeficiency; CeD, celiac disease; SIgAD, selective IgA deficiency; GFD, gluten-free diet; GVH, graft-versus-host; IVIG, intravenous immunoglobulin; IELs, intraepithelial lymphocytes; antiTG2, anti-tissue transglutaminase; EMA, endomysial antibodies; DGP, deamidated glutin peptides; GI, gastrointestinal.
2.3. Inflammatory Bowel Disease in Selective IgA Deficiency
Various authors seem to agree that the prevalence of IBD in SIgAD patients is increased in contrast to non-SIgAD patients. Ludvigsson et al., in a large population-based matched cohort study, reported a prevalence of 3.9% among SIgAD individuals to 0.81% among controls [12]. In research conducted by Azizi et al., the prevalence of IBD in SIgAD patients was 3.3% [40]. Both results were statistically significant. When concerning the coexistence of SIgAD and UC or SIgAD and CD separately, the prevalence was reported as 0.63% to 7.9% (weighted average of 1.73%) and between 1.21% and 15.8% (weighted average of 2.49%), respectively, in a comprehensive review of the literature performed by Odineal and Gershwin [41]. In the aforementioned Ludvigsson cohort study, the prevalence of UC in SIgAD patients was 1.7% in contrast to 0.46% in the control group. In the same study, the prevalence of CD in SIgAD individuals was 2.4%, compared to 0.42% among controls. Both results were statistically significant, suggesting an association between SIgAD and UC, as well as between SIgAD and CD [12].
To date, the literature indicates no significant differences between IBD in SIgAD and only IBD in terms of histological features, symptoms, and treatment. Histological findings in coexistent SIgAD and UC or CD are consistent with those concerning UC or CD separately [6][42].
2.4. Inflammatory Bowel Disease in Common Variable Immunodeficiency
Enteropathy is a common manifestation of CVID [28]. It is difficult to determine the actual prevalence of IBD among CVID patients. Not many studies on this topic are available and authors usually limit themselves to
stating that concomitant CVID and IBD occurrence exceeds this in the general population. In many reviews, the prevalence of 2–13% of IBD among CVID patients repeats [34][43][44]. Most authors agree on that the clinical signs in IBD or IBD-like within CVID patients show no significant differences with those without CVID and include weight loss, chronic diarrhoea, rectal bleeding, abdominal pain, and malabsorption [4][25][26][45]. The main difference concerns the possibility of co-occurring severe infections in immunocompromised patients [46]. Histopathology assessment of colon specimens from CVID patients shows endoscopic and histopathological features that overlap considerably with CD or UC [26][34][47][48]. One difference that can help distinguish idiopathic IBD from CVID enteropathy/colitis remains the paucity of plasma cells in their biopsy (observed in 68% of the patients) [34][49]. Many authors are in favour of the theory that CVID enteropathy/colitis is a distinct independent disorder, separate from classic IBD, as there is an aforementioned lack of plasma cells in the mucosa [50][51][52]. This approach highlights that CVID can mimic lymphocytic colitis, collagenous colitis, and colitis associated with graft-versus-host disease [53][54][55]. All the above-described observations suggest that some cases of gut inflammation in CVID patients can be diagnosed as true IBD, while others should be referred to as IBD-like. There is an ongoing discussion on this topic and no consensus has been reached yet.
The clinical approach in managing IBD in primary antibody deficiencies remains a challenge. Regardless of the doubts of whether IBD in CVID should be referred to as IBD, or rather IBD-like disease, or CVID enteropathy/colitis, most authors recommend using therapy schemes for IBD, with increased caution concerning immunosuppressive drugs [4][56]. However, there is little data about the therapeutical approach in this case. Gut inflammation in CVID is usually difficult to control and often resistant to standard IBD therapy [56][57]. Treatment of non-infectious GI disease in CVID includes corticosteroids, elimination of bacterial overgrowth with antibiotics, 5-aminosalicylic acid, 6-mercaptopurine, and azathioprine [56][58][56][59]. Besides, several groups reported substantial efficacy of targeted biological therapies, including anti-TNF-α drugs (infliximab and adalimumab), and the anti-IL−12/IL−23 monoclonal antibody ustekinumab [49][60][49][61][62][63][64][65]. Vedolizumab, an inhibitor of α4β7integrin, was also used in CVID-enteropathy, although with different results [28][66]. Furthermore, CVID patients with substantial T-cell defects require careful monitoring for fungal infections when on biological treatment. Immunoglobulin replacement therapy lowers the frequency of recurrent or severe infections and reduces the rate of hospitalization, but it was found to be ineffective in IBD-like disease in CVID [29][32][44].
A graphical summary concerning IBD and CVID is presented in Figure 2.
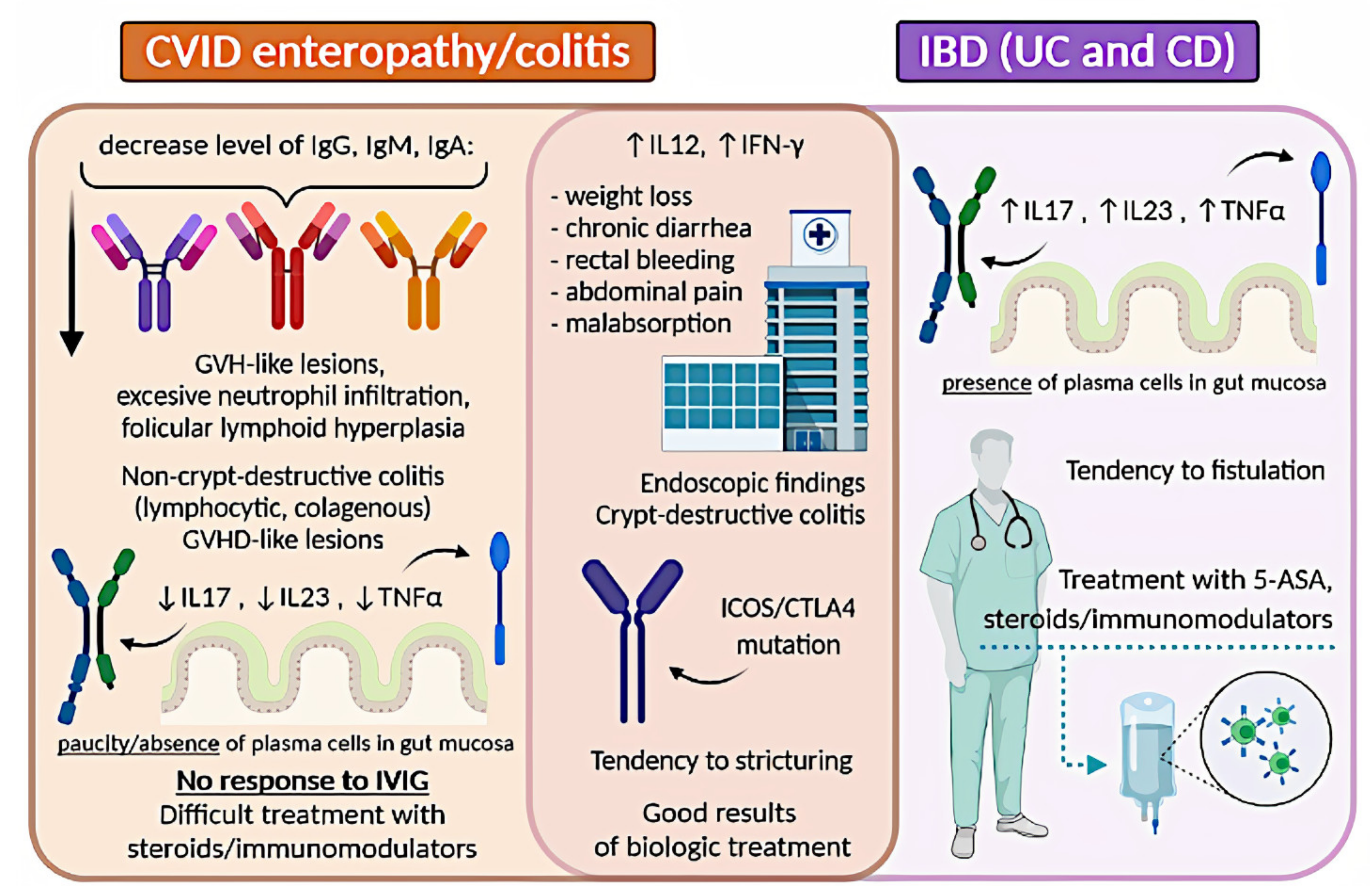
Figure 2. Comparison of IBD and CVID enteropathy/colitis. CVID, common variable immunodeficiency; IBD, inflammatory bowel disease; UC, ulcerative colitis; CD, Crohn’s disease; GVH, graft-versus-host; IL, interleukin; TNF-α, tumor necrosis factor-alpha; IVIG, intravenous immunoglobulin; INF-γ, interferon-gamma; 5-ASA, 5-aminosalicylic acid.
References
- Natalie A. Molodecky; Ing Shian Soon; Doreen M. Rabi; William A. Ghali; Mollie Ferris; Greg Chernoff; Eric I Benchimol; Remo Panaccione; Subrata Ghosh; H.W Barkema; et al.Gilaad G Kaplan Increasing Incidence and Prevalence of the Inflammatory Bowel Diseases With Time, Based on Systematic Review. Gastroenterology 2012, 142, 46-54.e42, 10.1053/j.gastro.2011.10.001.
- Alberto Rubio–Tapia; Robert A. Kyle; Edward L. Kaplan; Dwight R. Johnson; William Page; Frederick Erdtmann; Tricia L. Brantner; W. Ray Kim; Tara K. Phelps; Brian D. Lahr; et al.Alan R. ZinsmeisterL. Joseph MeltonJoseph A. Murray Increased Prevalence and Mortality in Undiagnosed Celiac Disease. Gastroenterology 2009, 137, 88-93, 10.1053/j.gastro.2009.03.059.
- Benjamin Lebwohl; David S Sanders; Peter H R Green; Coeliac disease. The Lancet 2018, 391, 70-81, 10.1016/s0140-6736(17)31796-8.
- Shradha Agarwal; Charlotte Cunningham-Rundles; Gastrointestinal Manifestations and Complications of Primary Immunodeficiency Disorders.. Immunology and Allergy Clinics of North America 2019, 39, 81-94, 10.1016/j.iac.2018.08.006.
- Gholamreza Azizi; Vahid Ziaee; Marzieh Tavakol; T. Alinia; R. Yazdai; Hamed Mohammadi; Hassan Abolhassani; Asghar Aghamohammadi; Approach to the Management of Autoimmunity in Primary Immunodeficiency. Scandinavian Journal of Immunology 2017, 85, 13-29, 10.1111/sji.12506.
- David Schwimmer; Sarah Glover; Primary Immunodeficiency and the Gut. Gastroenterology Clinics of North America 2019, 48, 199-220, 10.1016/j.gtc.2019.02.004.
- Vicki Modell; Jordan S. Orange; Jessica Quinn; Fred Modell; Global report on primary immunodeficiencies: 2018 update from the Jeffrey Modell Centers Network on disease classification, regional trends, treatment modalities, and physician reported outcomes. Immunologic Research 2018, 66, 367-380, 10.1007/s12026-018-8996-5.
- Hassan Abolhassani Md; Nima Rezaei; Payam Mohammadinejad; Babak Mirminachi; Lennart Hammarström Md; Asghar Aghamohammadi; Important differences in the diagnostic spectrum of primary immunodeficiency in adults versus children. Expert Review of Clinical Immunology 2015, 11, 289-302, 10.1586/1744666x.2015.990440.
- Abdulbaqi Al-Toma; Umberto Volta; Renata Auricchio; Gemma Castillejo; David S Sanders; Christophe Cellier; Chris J Mulder; Knut E A Lundin; European Society for the Study of Coeliac Disease (ESsCD) guideline for coeliac disease and other gluten-related disorders.. United European Gastroenterology Journal 2019, 7, 583-613, 10.1177/2050640619844125.
- Kelly E. McGowan; Martha E. Lyon; J. Decker Butzner; Celiac Disease and IgA Deficiency: Complications of Serological Testing Approaches Encountered in the Clinic. Clinical Chemistry 2008, 54, 1203-1209, 10.1373/clinchem.2008.103606.
- Karmtej Singh; Christopher Chang; M. Eric Gershwin; IgA deficiency and autoimmunity. Autoimmunity Reviews 2014, 13, 163-177, 10.1016/j.autrev.2013.10.005.
- Jonas F Ludvigsson; Martin Neovius; Lennart Hammarström; Association Between IgA Deficiency & Other Autoimmune Conditions: A Population-Based Matched Cohort Study. Journal of Clinical Immunology 2014, 34, 444-451, 10.1007/s10875-014-0009-4.
- A. Lenhardt; A. Plebani; Federico Marchetti; T. Gerarduzzi; Tarcisio Not; A. Meini; V. Villanacci; S. Martelossi; Alessandro Ventura; Role of human-tissue transglutaminase IgG and anti-gliadin IgG antibodies in the diagnosis of coeliac disease in patients with selective immunoglobulin A deficiency. Digestive and Liver Disease 2004, 36, 730-734, 10.1016/j.dld.2004.06.017.
- Ning Wang; Lennart Truedsson; Kerstin Elvin; Bengt Å. Andersson; Johan Rönnelid; Lucia Mincheva-Nilsson; Annica Lindkvist; Jonas F. Ludvigsson; Lennart Hammarström; Charlotte Dahle; et al. Serological Assessment for Celiac Disease in IgA Deficient Adults. PLOS ONE 2014, 9, e93180, 10.1371/journal.pone.0093180.
- Kumar Pallav; Hua Xu; Daniel A. Leffler; Toufic Kabbani; Ciaran P. Kelly; Immunoglobulin A deficiency in celiac disease in the United States.. Journal of Gastroenterology and Hepatology 2016, 31, 133-7, 10.1111/jgh.13176.
- Harry E. Prince; Gary L. Norman; Walter L. Binder; Immunoglobulin A (IgA) Deficiency and Alternative Celiac Disease-Associated Antibodies in Sera Submitted to a Reference Laboratory for Endomysial IgA Testing. Clinical Diagnostic Laboratory Immunology 2000, 7, 192-196, 10.1128/cdli.7.2.192-196.2000.
- Ingrid Dahlbom; Martin Olsson; Nahal Kazemi Forooz; Anders G. Sjöholm; Lennart Truedsson; Tony Hansson; Immunoglobulin G (IgG) Anti-Tissue Transglutaminase Antibodies Used as Markers for IgA-Deficient Celiac Disease Patients. Clinical Diagnostic Laboratory Immunology 2005, 12, 254-258, 10.1128/cdli.12.2.254-258.2005.
- Danilo Villalta; Maria Grazia Alessio; Marilina Tampoia; Elio Tonutti; Ignazio Brusca; Marcello Bagnasco; Giampaola Pesce; Sergio Stella; Nicola Bizzaro; Testing for IgG class antibodies in celiac disease patients with selective IgA deficiency.. Clinica Chimica Acta 2007, 382, 95-99, 10.1016/j.cca.2007.03.028.
- Amir H Abdul Latiff; Michael A Kerr; The clinical significance of immunoglobulin A deficiency.. Annals of Clinical Biochemistry: International Journal of Laboratory Medicine 2007, 44, 131-139, 10.1258/000456307780117993.
- Enrico Valletta; M Fornaro; S Pecori; G Zanoni; Selective immunoglobulin A deficiency and celiac disease: let's give serology a chance.. Journal of Investigational Allergology and Clinical Immunology 2011, 21, 242–244.
- J Winkelstein; The X-Linked Hyper-IgM Syndrome: Clinical and Immunologic Features of 79 Patients. The Pediatric Infectious Disease Journal 2004, 23, 983, 10.1097/01.inf.0000137582.89107.f4.
- R. Fuleihan; N. Ramesh; R. Loh; H. Jabara; R. S. Rosen; T Chatila; S. M. Fu; I. Stamenkovic; R. S. Geha; Defective expression of the CD40 ligand in X chromosome-linked immunoglobulin deficiency with normal or elevated IgM.. Proceedings of the National Academy of Sciences 1993, 90, 2170-2173, 10.1073/pnas.90.6.2170.
- Emily A. Leven; Patrick Maffucci; Hans D. Ochs; Paul R. Scholl; Rebecca H. Buckley; Ramsay L. Fuleihan; Raif S. Geha; Coleen K. Cunningham; Francisco A. Bonilla; Mary Ellen Conley; et al.Ronald M. FerdmanVivian Hernandez-TrujilloJennifer M. PuckKathleen SullivanElizabeth A. SecordManish RameshCharlotte Cunningham-Rundles Hyper IgM Syndrome: a Report from the USIDNET Registry. Journal of Clinical Immunology 2016, 36, 490-501, 10.1007/s10875-016-0291-4.
- Charlotte Cunningham-Rundles; Physiology of IgA and IgA deficiency.. Journal of Clinical Immunology 2001, 21, 303-309, 10.1023/a:1012241117984.
- Shradha Agarwal; Lloyd Mayer; Diagnosis and Treatment of Gastrointestinal Disorders in Patients With Primary Immunodeficiency. Clinical Gastroenterology and Hepatology 2013, 11, 1050-1063, 10.1016/j.cgh.2013.02.024.
- Jason A. Daniels; Howard M. Lederman; Anirban Maitra; Elizabeth A. Montgomery; Gastrointestinal Tract Pathology in Patients With Common Variable Immunodeficiency (CVID). American Journal of Surgical Pathology 2007, 31, 1800-1812, 10.1097/pas.0b013e3180cab60c.
- Alberto Rubio-Tapia; Ivor D Hill; Ciarán P Kelly; Audrey H. Calderwood; Joseph A. Murray; ACG Clinical Guidelines: Diagnosis and Management of Celiac Disease. American Journal of Gastroenterology 2013, 108, 656-676, 10.1038/ajg.2013.79.
- Mathieu Uzzan; Huaibin M. Ko; Saurabh Mehandru; C. Cunningham-Rundles; Gastrointestinal Disorders Associated with Common Variable Immune Deficiency (CVID) and Chronic Granulomatous Disease (CGD).. Current Gastroenterology Reports 2016, 18, 17-17, 10.1007/s11894-016-0491-3.
- C. Cunningham-Rundles; Common variable immune deficiency: Dissection of the variable. Immunological Reviews 2018, 287, 145-161, 10.1111/imr.12728.
- Yuan, Q.; Bousvaros, A. Chapter 38—Gastrointestinal manifestations of primary immunodeficiency. In Pediatric Gastrointestinal and Liver Disease, 3rd ed.; Wyllie, R., Hyams, J.S., Kay, M., Eds.; W.B. Saunders: London, UK, 2006; pp. 601–616. ISBN 978-0-7216-3924-6.
- Neslihan Edeer Karaca; Ezgi Ulusoy Severcan; Burcu Guven Bilgin; Elif Azarsiz; Sanem Akarcan; Nursen Cigerci Gunaydın; Nesrin Gulez; Ferah Genel; Guzide Aksu; Necil Kutukculer; et al. Familial inheritance and screening of first-degree relatives in common variable immunodeficiency and immunoglobulin A deficiency patients. International Journal of Immunopathology and Pharmacology 2018, 32, 2058738418779458, 10.1177/2058738418779458.
- Georgia Malamut; Virginie Verkarre; Felipe Suarez; Jean-François Viallard; Anne-Sophie Lascaux; Jacques Cosnes; Yoram Bouhnik; Olivier Lambotte; Dominique Béchade; Marianne Ziol; et al.Anne LavergneOlivier HermineNadine Cerf-BensussanChristophe Cellier The Enteropathy Associated With Common Variable Immunodeficiency: The Delineated Frontiers With Celiac Disease. American Journal of Gastroenterology 2010, 105, 2262-2275, 10.1038/ajg.2010.214.
- Giuseppe Luzi; Angelo Zullo; Filippo Iebba; Vittorio Rinaldi; Lupe Sanchez Mete; Maurizio Muscaritoli; Fernando Aiuti; Duodenal pathology and clinical-immunological implications in common variable immunodeficiency patients. American Journal of Gastroenterology 2003, 98, 118-121, 10.1016/s0002-9270(02)05827-6.
- Stella Hartono; Michelina Rosa Ippoliti; Maria Mastroianni; Ricardo Torres; Nicholas L. Rider; Gastrointestinal Disorders Associated with Primary Immunodeficiency Diseases. Clinical Reviews in Allergy & Immunology 2018, 57, 145-165, 10.1007/s12016-018-8689-9.
- Nils Venhoff; Florian Emmerich; Michaela Neagu; Ulrich Salzer; Corinna Koehn; Sibyll Driever; Wolfgang Kreisel; Marta Rizzi; Nora M. Effelsberg; Florian Kollert; et al.Sigune GoldackerReinhard E. VollKlaus WarnatzJens Thiel The Role of HLA DQ2 and DQ8 in Dissecting Celiac-Like Disease in Common Variable Immunodeficiency. Journal of Clinical Immunology 2013, 33, 909-916, 10.1007/s10875-013-9892-3.
- Ahmed Dawood Mohammed; A. Wadud Khan; Ioulia Chatzistamou; Douja Chamseddine; Katie Williams-Kang; Mason Perry; Reilly Enos; Angela Murphy; Gregorio Gomez; Ahmed Aladhami; et al.Carole A. OskeritzianAmy JollyYan ChangShuqian HeZui PanJason Lee Kubinak Gut Antibody Deficiency in a Mouse Model of CVID Results in Spontaneous Development of a Gluten-Sensitive Enteropathy. Frontiers in Immunology 2019, 10, 2484, 10.3389/fimmu.2019.02484.
- S F Jørgensen; Henrik M. Reims; Pål Aukrust; Knut E A Lundin; Børre Fevang; CVID and Celiac Disease. American Journal of Gastroenterology 2017, 112, 393-393, 10.1038/ajg.2016.551.
- Federico Biagi; Paola I. Bianchi; Alessandra Zilli; Alessandra Marchese; Ombretta Luinetti; Vassilios Lougaris; Alessandro Plebani; Vincenzo Villanacci; Gino Roberto Corazza; The Significance of Duodenal Mucosal Atrophy in Patients With Common Variable Immunodeficiency. American Journal of Clinical Pathology 2012, 138, 185-189, 10.1309/ajcpeiilh2c0wfye.
- Sampsa Pikkarainen; Timi Martelius; Ari Ristimäki; Sanna Siitonen; Mikko Seppänen; M.A. Färkkilä; A High Prevalence of Gastrointestinal Manifestations in Common Variable Immunodeficiency. American Journal of Gastroenterology 2019, 114, 648-655, 10.14309/ajg.0000000000000140.
- Gholamreza Azizi; Marzieh Tavakol; H Rafiemanesh; Fatemeh Kiaee; Reza Yazdani; Amin Heydari; Kosar Abouhamzeh; Pardis Anvari; Sara Mohammadikhajehdehi; Laleh Sharifia; et al.Yasser BagheriHamed MohammadiHassan AbolhassaniAsghar Aghamohammadi Autoimmunity in a cohort of 471 patients with primary antibody deficiencies. Expert Review of Clinical Immunology 2017, 13, 1099-1106, 10.1080/1744666x.2017.1384312.
- David Odineal; M. Eric Gershwin; The Epidemiology and Clinical Manifestations of Autoimmunity in Selective IgA Deficiency. Clinical Reviews in Allergy & Immunology 2019, 58, 107-133, 10.1007/s12016-019-08756-7.
- Yuki Asada; Hajime Isomoto; Saburo Shikuwa; Chun Yang Wen; Eiichiro Fukuda; Masaru Miyazato; Kenta Okamoto; Takashi Nakamura; Hitoshi Nishiyama; Yohei Mizuta; et al.Kiyoshi MigitaMasahiro ItoShigeru Kohno Development of ulcerative colitis during the course of rheumatoid arthritis: Association with selective IgA deficiency. World Journal of Gastroenterology 2006, 12, 5240-5243, 10.3748/wjg.v12.i32.5240.
- Shradha Agarwal; Lloyd Mayer; Gastrointestinal manifestations in primary immune disorders. Inflammatory Bowel Diseases 2010, 16, 703-711, 10.1002/ibd.21040.
- Shradha Agarwal; Paul Smereka; Noam Harpaz; Charlotte Cunningham-Rundles; Lloyd Mayer; Characterization of immunologic defects in patients with common variable immunodeficiency (CVID) with intestinal disease. Inflammatory Bowel Diseases 2011, 17, 251-259, 10.1002/ibd.21376.
- Elena S. Resnick; Erin L. Moshier; James H. Godbold; Charlotte Cunningham-Rundles; Morbidity and mortality in common variable immune deficiency over 4 decades. Blood 2012, 119, 1650-1657, 10.1182/blood-2011-09-377945.
- Daniel Tegtmeyer; Maximilian Seidl; Patrick Gerner; Ulrich Baumann; Christian Klemann; Inflammatory bowel disease caused by primary immunodeficiencies-Clinical presentations, review of literature, and proposal of a rational diagnostic algorithm. Pediatric Allergy and Immunology 2017, 28, 412-429, 10.1111/pai.12734.
- Brian P. Bosworth; Abraham Sanders; Charles Maltz; Common variable immunodeficiency masquerading as Crohnʼs ileocolitis. Inflammatory Bowel Diseases 2006, 12, 151-152, 10.1097/01.mib.0000200344.61707.9c.
- Jonas Mudter; Stefan Wirtz; Benno Weigmann; Imke Tiede; Ingrid Tubbe; Ralf Kiesslich; Peter R. Galle; Hans A. Lehr; Markus F. Neurath; Crohn’s-like Colitis in a Patient with Immunodeficiency Associated with a Defect in Expression of Inducible Costimulator. Digestive Diseases and Sciences 2006, 51, 711-717, 10.1007/s10620-006-3196-x.
- Juan María Vazquez Morón; Héctor Pallarés-Manrique; Ignacio Javier Martín-Suárez; Beatriz Benítez-Rodríguez; Manuel Ramos-Lora; Crohn's-like disease in a patient with common variable immunodeficiency treated with azathioprine and adalimumab.. Revista Española de Enfermedades Digestivas 2013, 105, 299-302, 10.4321/s1130-01082013000500010.
- Ishaan Kalha; Joseph H. Sellin; Common variable immunodeficiency and the gastrointestinal tract. Current Gastroenterology Reports 2004, 6, 377-383, 10.1007/s11894-004-0053-y.
- Ahmad Khodadad; Asghar Aghamohammadi; Nima Parvaneh; Nima Rezaei; Fatemeh Mahjoob; Mohammad Bashashati; Masoud Movahedi; Mohammad Reza Fazlollahi; Fariborz Zandieh; Zahra Roohi; et al.Sina AbdollahzadeAli SalavatiAli KouhiBahram TalebpourNasser Ebrahimi Daryani Gastrointestinal Manifestations in Patients with Common Variable Immunodeficiency. Digestive Diseases and Sciences 2007, 52, 2977-2983, 10.1007/s10620-006-9736-6.
- Nil Comunoglu; Sinem Kara; Nuray Kepil; Inflammatory bowel disease-like colitis pathology in a patient with common variable immune deficiency. BMJ Case Reports 2015, 2015, bcr2014207177, 10.1136/bcr-2014-207177.
- K Washington; T T Stenzel; R H Buckley; M R Gottfried; Gastrointestinal pathology in patients with common variable immunodeficiency and X-linked agammaglobulinemia.. The American Journal of Surgical Pathology 1996, 20, 1240-52.
- Michael F. Byrne; Derval Royston; Stephen E Patchett; Association of common variable immunodeficiency with atypical collagenous colitis. European Journal of Gastroenterology & Hepatology 2003, 15, 1051-1053, 10.1097/00042737-200309000-00019.
- G. Castellano; D. Moreno; O. Galvao; C. Ballestín; F. Colina; Manuela Mollejo; J. D. Morillas; J. A. Solís Herruzo; Malignant Lymphoma of Jejunum with Common Variable Hypogammaglobulinemia and Diffuse Nodular Hyperplasia of the Small Intestine. Journal of Clinical Gastroenterology 1992, 15, 128-135, 10.1097/00004836-199209000-00010.
- Antonio Pecoraro; Liliana Nappi; Ludovica Crescenzi; Francesco P. D’Armiento; Arturo Genovese; Giuseppe Spadaro; Chronic Diarrhea in Common Variable Immunodeficiency: a Case Series and Review of the Literature. Journal of Clinical Immunology 2017, 38, 67-76, 10.1007/s10875-017-0461-z.
- M Sanges; G Spadaro; M Miniero; D Mattera; R Sollazzo; F P D'armiento; Giovanni Domenico De Palma; A Pecoraro; F Borrelli; A Genovese; et al.A. D'arienzo Efficacy of subcutaneous immunoglobulins in primary immunodeficiency with Crohn's-like phenotype: report of a case.. European review for medical and pharmacological sciences 2015, 19, 2641-5.
- Shradha Agarwal; Lloyd Mayer; Pathogenesis and treatment of gastrointestinal disease in antibody deficiency syndromes. Journal of Allergy and Clinical Immunology 2009, 124, 658-664, 10.1016/j.jaci.2009.06.018.
- Najwa Elnachef; Marc McMorris; William D. Chey; Successful Treatment of Common Variable Immunodeficiency Disorder-Associated Diarrhea With Budesonide: A Case Report. American Journal of Gastroenterology 2007, 102, 1322-1325, 10.1111/j.1572-0241.2007.01141.x.
- Jose G. Ruiz De Morales; Fernando Muñoz; Mercedes Hernando; Successful Treatment of Common Variable Immunodeficiency-associated Inflammatory Bowel Disease With Ustekinumab. Journal of Crohn's and Colitis 2017, 11, 1154-1155, 10.1093/ecco-jcc/jjx024.
- Cátia Arieira; Francisca Dias De Castro; Maria João Moreira; José Cotter; Common Variable Immunodeficiency-Associated Inflammatory Enteropathy: The New Era of Biological Therapy.. GE - Portuguese Journal of Gastroenterology 2018, 25, 322-326, 10.1159/000486448.
- I. Chua; R. Standish; S. Lear; M. Harbord; E. Eren; M. Raeiszadeh; S. Workman; D. Webster; Anti-tumour necrosis factor-α therapy for severe enteropathy in patients with common variable immunodeficiency (CVID). Clinical & Experimental Immunology 2007, 150, 306-311, 10.1111/j.1365-2249.2007.03481.x.
- Pilar Nos; G. Bastida; Belén Beltrán; Mariam Aguas; Julio Ponce; Crohn's Disease in Common Variable Immunodeficiency: Treatment with Antitumor Necrosis Factor Alpha. American Journal of Gastroenterology 2006, 101, 2165-2166, 10.1111/j.1572-0241.2006.00763_5.x.
- Yuko Akazawa; Fuminao Takeshima; Hiroyuki Yajima; Daisuke Imanishi; Tsutomu Kanda; Kayoko Matsushima; Hitomi Minami; Naoyuki Yamaguchi; Ken Ohnita; Hajime Isomoto; et al.Tomayoshi HayashiMasahiro NakashimaKazuhiko Nakao Infliximab therapy for Crohn’s-like disease in common variable immunodeficiency complicated by massive intestinal hemorrhage: a case report. BMC Research Notes 2014, 7, 382-382, 10.1186/1756-0500-7-382.
- Cristina Saldaña Dueñas; Saioa Rubio Iturria; Immmunodeficiencies and autoimmune diseases: Common variable immunodeficiency and Crohn-like. Revista Española de Enfermedades Digestivas 2015, 108, 520-3, 10.17235/reed.2015.3872/2015.
- Brigid Boland; Marc A. Riedl; Mark A. Valasek; Sheila E. Crowe; William J. Sandborn; Vedolizumab in Patients With Common Variable Immune Deficiency and Gut Inflammation. American Journal of Gastroenterology 2017, 112, 1621-1621, 10.1038/ajg.2017.246.