Lignin biodegradation has been extensively studied in white-rot fungi, which largely belong to order Polyporales. Among the enzymes that wood-rotting polypores secrete, lignin peroxidases (LiPs) have been labeled as the most efficient. A recent thorough study of 52 Agaricomycetes genomes has revealed the high presence of putative ligninolytic peroxidases in fungi belonging to the order Agaricales. These include the first LiP outside the order Polyporales, identified in the genome of the mushroom Agrocybe pediades (ApeLiP) as a case of parallel and convergent evolution of LiPs between Agaricales and Polyporales.
- Agaricales
- lignin peroxidase
- catalytic tryptophan
- crystal structure
- transient-state kinetics
- reduction potential
- model dimers
- non-phenolic lignin
- lignosulfonate degradation
- NMR spectroscopy
1. Introduction
Plant biomass is an abundant renewable resource that has attracted an increasing interest during the last decades in the context of lignocellulose biorefinery[1] [2][1,2]. It is largely made up of the polysaccharides cellulose and hemicellulose, and the aromatic polymer lignin [3]. While cellulose is used in the pulp and paper industry and in the production of added value compounds and biofuels, lignin has been underused, mainly due to its recalcitrance[4] [5][4,5]. For this reason, organisms able to modify lignin in nature have been widely studied in the last decades, either for a greener degradation of the polymer or in the production of added-value aromatic compounds [6][7][8][9][10][6,7,8,9,10].
Although some bacteria[11][12] [11,12] and soft rot fungi[13] [13] are also involved in lignin degradation acting on the phenolic moiety and degradation products, white-rot fungi are the only organisms able to extensively mineralize native lignin[9][14][15] [9,14,15]. They use an array of oxidative enzymatic tools for its extracellular degradation, among which the ligninolytic peroxidases play a major role[9][16] [9,16]. These enzymes are part of class-II of the peroxidase–catalase superfamily [17] and, according to the oxidation sites in their molecular architecture, are classified into three families[16] [16]: (i) manganese peroxidases (MnPs), that harbor a Mn-binding site where Mn2+ is oxidized to Mn3+. This manganese cation acts as a diffusible mediator both oxidizing the minor phenolic moiety of lignin and generating strong oxidizers by initiating lipid peroxidation[18] [18]. Also, members of the short MnP subfamily are able to oxidize phenolic compounds in direct contact with the heme cofactor[19] [19]; (ii) lignin peroxidases (LiPs), which contain a solvent-exposed catalytic tryptophan where they directly oxidize high redox-potential non-phenolic aromatic substrates[20][21] [20,21]; and (iii) versatile peroxidases (VPs), that accommodate in a single enzyme the catalytic sites previously described for MnPs and LiPs [22]. In the evolution of Polyporales, where most white-rot fungi are included, MnPs are ancient enzymes from which the other two families arose, first by incorporating a solvent-exposed tryptophan generating VPs, and later by losing the Mn-binding site generating the LiPs that we can find today together with more ancient families [19][23][19,23].
LiPs are the most efficient enzymes oxidizing lignin, being isoenzyme H8 from the white-rot fungus Phanerochaete chrysosporium [16] corresponding to LiPA from its sequenced genome [24], the best known ligninolytic peroxidase. This enzyme has been extensively studied from different viewpoints, demonstrating its capacity to oxidize non-phenolic lignin directly[25][26][27] [25,26,27]. Since its discovery [28], different LiP and LiP-like enzymes have been characterized (see family AA2 in CAZY database, http://www.cazy.org/AA2.html; accessed on 16 August 2021), but only in white-rot Polyporales. However, the recent investigation of dozens of genomes in species of Agaricales have revealed that LiP genes also appeared in this fungal order, through an evolutionary pathway that is different from the one that gave rise to the Polyporales LiPs [29]. These include the LiP of the grass-litter mushroom Agrocybe pediades (ApeLiP herein after), whose main similarities and differences with other reported ligninolytic peroxidases are discussed below.
2. ApeLiP Activation and LRET Oxidation of Lignin Models
The successfull crystallization of the protein allowed to discern the main structural similarities of this LiP with other fungal ligninolytic peroxidases (Figure 1)[30] [62]. Thus, the heme cofactor was found buried in the structure and sandwiched by two alpha helices which contained conserved residues involved in the heterolytic cleavage of H2O2 (in the distal side of the heme pocket) and in the modulation of the redox-potential of the enzyme (in the proximal side, with a histidine residue acting as fifth ligand of the heme iron). Therefore, it was expected to observe a similar enzyme activation by H2O2 to other ligninolytic peroxidases. In fact, its apparent second-order rate constant for CI formation, (4.7 ± 0.01) × 106 s−1·M−1, was in the same range of Pleurotus eryngii VP[31] [43], P. chrysosporium MnP[32] [42] and Trametes cervina LiP [33][63], although this rate was one order of magnitude higher than the reported for P. chrysosporium LiPA (isoenzyme H8)[34] [64]. These differences in the reaction rate with H2O2 could affect the oxidative stability of peroxidases, as confirmed by a VP variant at the heme distal side showing slower activation by H2O2 and enhanced oxidative stability[35] [65].
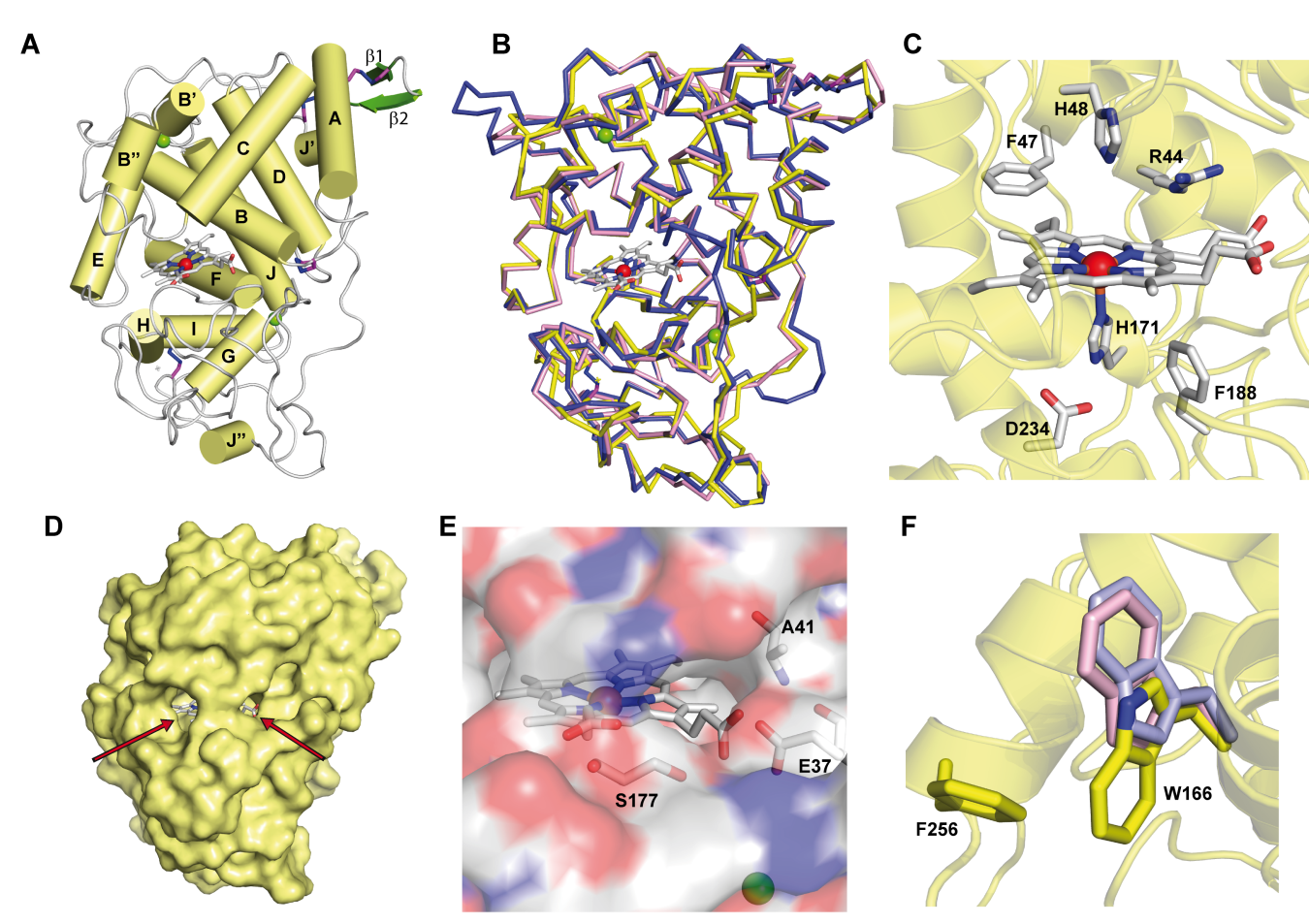
Figure 1. Crystal structure of ApeLiP. A) Scheme of the overall structure showing 14 α-helices (yellow cylinders) and two small antiparallel β-strands (green), the heme cofactor (CPK sticks) with the iron center (red sphere), two structural calcium ions (green spheres), and four disulphide bonds (purple-blue sticks) (PDB entry 7OO5). B) Structural alignment of the ApeLiP backbone (yellow) with the backbones of P. chrysosporium LiPA (blue) and P. eryngii VPL (magenta) (from PDB entries 1B82 and 2BOQ, respectively) (heme and structural calcium ions are shown). C) Heme cavity typical from class-II peroxidases including His171, Asp234 and Phe188 at the proximal side (below the heme plane); and His48, Arg44 and Phe47 at the distal side (above the heme plane). D) Protein surface with indication (arrows) of two access channels to the heme cofactor. E) Small access channel located on the heme propionates lacking a Mn2+ binding site, with Ala41, Glu37 and Ser177 occupying the positions of the cation ligands in MnP and VP enzymes. F) Superimposition of ApeLiP (yellow sticks), P. chrysosporium LiPA (pale violet), and P. eryngii VPL (pale pink) catalytic tryptophans.
The crystal structure also revealed the presence of a putative oxidation site at the protein surface that includes a solvent-exposed tryptophan. This tryptophan, Trp166 in ApeLiP, is conserved in most LiPs and VPs and would be involved in the oxidation of high redox-potential and bulky molecules, including lignin [21][26][27][21,26,27], by long-range electron transfer (LRET) from the protein surface to the activated heme cofactor[36] [66]. To evaluate this hypothesis, the catalytic properties of the wild-type enzyme and its tryptophan-less W166A variant were analyzed using different phenolic and non-phenolic aromatic compounds. Thereby, ApeLiP was able to oxidize the non-phenolic lignin model dimer veratrylglycerol-β-guaiacyl ether (VGE) and also veratryl alcohol (VA). Moreover, this enzyme oxidizes high redox-potential dyes such as Reactive Black 5 (RB5) without the requirement of redox mediators, a feature only observed in VPs[37][38] [43,67] and ancestral LiPs[23][39][40] [23,44,68], but not in P. chrysosporium LiPA and Bjerkandera adusta LiP2, that require VA for efficient oxidation of different aromatic compounds and dyes[41][42][43] [69,70,71]. As no activity was observed when the W166A mutation was present, the importance of this residue in degradation of non-phenolic lignin and other recalcitrant molecules by ApeLiP stands clear.
Moreover, phenolic lignin model compounds such as guaiacylglycerol-β-guaiacyl ether (GGE) and 2,6-dimethoxyphenol (DMP), and the low-redox-potential dye 2,2'-azinobis(3-ethylbenzothiazoline-6-sulfonate) (ABTS) are oxidized with higher catalytic efficiencies than observed for other LiPs[19] [19] and VPs [32][41]. Besides, biphasic kinetics were observed for ABTS oxidation, indicating the presence of high and low efficiency oxidation sites[32] [41]. Disappearance of high-efficiency oxidation of phenolic compounds when Trp166 was removed suggests that this residue would also play a key role in the oxidation of phenolic lignin by ApeLiP.
The protein environment around the catalytic tryptophan was also evaluated as it can significantly affect substrate recognition and/or oxidation by LiPs and VPs. Thus, an electronegative environment would stabilize the VA cation radical to act as an enzyme-bound redox mediator[42] [72] at the same time that it can displace the VA oxidation reaction toward the oxidized forms. Moreover, acidic residues would lower the local pH, providing a higher redox-potential to the tryptophanyl radical[43] [73]. The less acidic environment of Trp166 comparing to that observed around Trp164 of P. chrysosporium LiP (Figure 2) could explain the lower oxidation power of ApeLiP on VA, with its activity and substrate recognition properties closer to those of VPs than of LiPs.
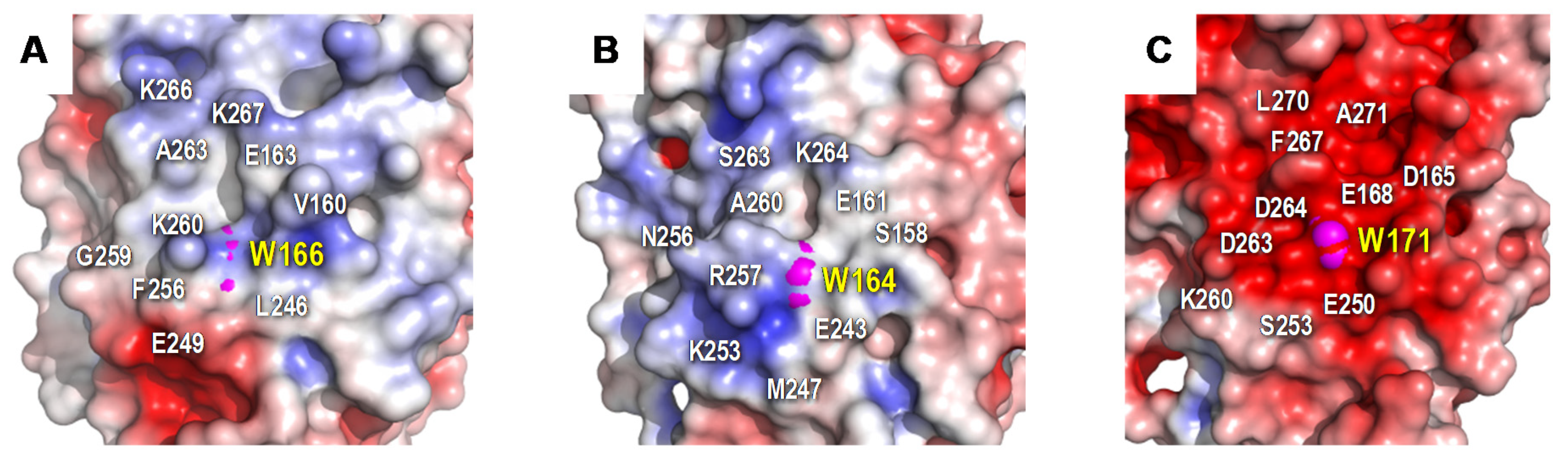
Figure 2. Electrostatic surface of the catalytic tryptophan environment in: A) ApeLiP, B) P. eryngii VPL, and C) P. chrysosporium LiPA. Shown is the positioning of catalytic tryptophan (pink spheres) and close acidic and basic residues (together with other surrounding amino acids). Negative (red) and positive (blue) charges are indicated. Based on crystal structure solved in the present work (A), and in PDB entries 2BOQ (B) and 1B82 (C).
3. Heme-Channel Oxidation Site
As reported for plant and fungal generic peroxidases and for P. eryngii VP, phenolic and other low redox-potential substrates can be oxidized directly by the heme through the channel that gives access to H2O2[44][45] [41,55]. However, the narrowness of this channel in most LiPs impedes these aromatic substrates to directly interact with the cofactor[30] [62]. The size and shape similarities between the ApeLiP and VP heme channels may explain the kinetic identification of a low-efficiency site for ABTS oxidation in the former enzyme, which remained when Trp166 was removed. Moreover, although the catalytic tryptophan was responsible for high-efficiency oxidation of DMP at pH 3, its optimal oxidation by the W166A variant was at pH 8, suggesting that this substrate may be oxidized by the heme cofactor under more basic conditions.
The ApeLiP ability to oxidize aromatic substrates (VGE included) at basic pH is a unique characteristic among the best known ligninolytic peroxidases, which are inactivated under these conditions due to Ca2+ loss and heme pocket collapse with hexacoordination of the heme iron [46][47][48][49][74,75,76,77]. Although the exact structural basis for this “basic peroxidase” activity remains to be identified, it seems related to direct substrate oxidation at the heme channel, in agreement with DMP oxidation results by the Trp-less ApeLiP variant. Catalytic activity at basic pH has been built in a VP by directed evolution, and the oxidation of low-redox potential substrates in direct contact with the heme cofactor was related with the stabilization of its heme pocket at alkaline pH[50][51] [78,79]. This suggests a higher stability of the heme and its environment in ApeLiP at basic pH compared to other ligninolytic peroxidases.
4. Intriguing Catalytic Cycle and Lignin Decay Abilities
The catalytic cycle of ligninolytic peroxidases starts with a two-electron oxidation of the resting state enzyme (RS) by H2O2, forming compound-I (CI). This intermediate is then reduced back to the RS enzyme via compound-II (CII) by two one-electron substrate oxidations in direct contact with the heme cofactor or at the solvent-exposed tryptophan. The stopped-flow spectrophotometric inspection of the ApeLiP catalytic cycle showed differences with respect to LiPs from Polyporales. The recently studied evolution of reduction potential in these ligninolytic peroxidases shows an increase through time for all the catalytic pairs, culminating in the highest E°’ of extant LiPs[39] [44]. However, ApeLiP showed comparatively low E°’ values. Those of its CI/RS, CI/CII and CII/RS pairs (Figure 3) follow the general tendency in other peroxidases, being E°’(CII/RS) the lowest (as corresponds to the rate-limiting step) and E°’(CI/CII) the highest (explaining the instability of CI). However, all the values for ApeLiP are up to 70 mV lower than the reduction potentials observed for LiPs of Polyporales. Additionally, the catalytic cycle of ApeLiP is intriguing. It was not possible to discriminate CI from CII using stopped-flow rapid spectrophotometry after ferrocyanide addition as classically used to generate peroxidase CII [52][53][80,81]. However, the putative intermediate forms the RS enzyme upon mixing with reducing substrate (either tyrosine or lignosulfonates) behaving as the typical CII of other ligninolytic peroxidases [39][54][44,59].
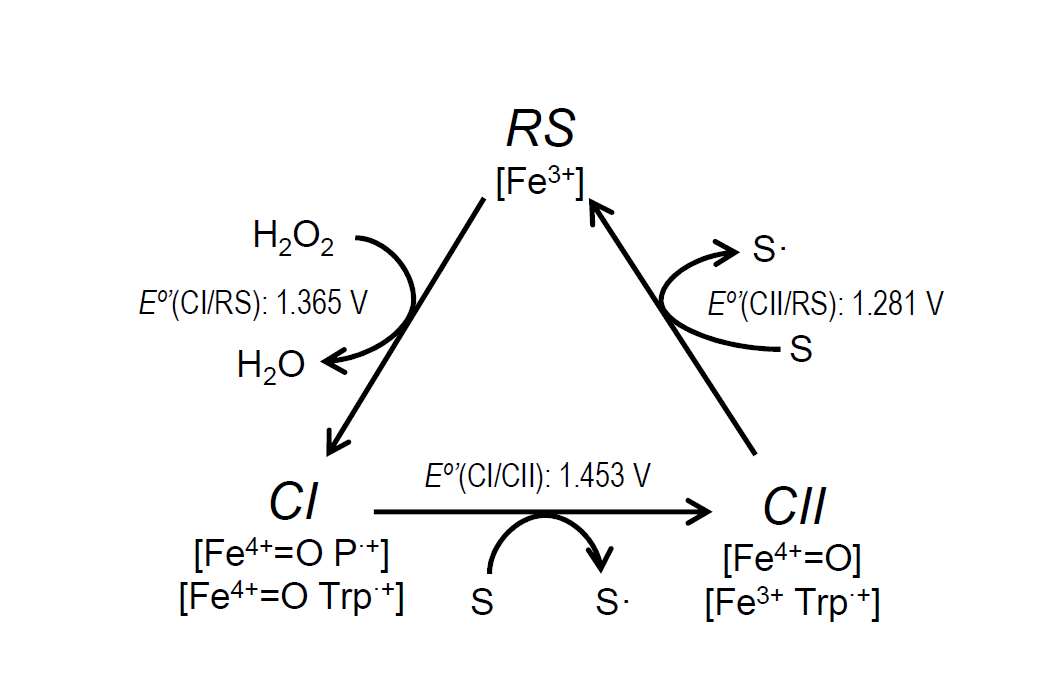
Figure 3. Catalytic cycle of ligninolytic peroxidases and E°’ of the ApeLiP CI/RS, CI/CII and CII/RS pairs estimated in the present study. The catalytic cycle includes: i) Resting state (RS; Fe3+) activation by H2O2 yielding CI, that retains two oxidizing equivalents in the form of Fe4+=O and porphyrin cation radical, P·+, or Fe4+=O and tryptophanyl cation radical, Trp·+; ii) CI reduction to CII, bearing only one oxidation equivalent on the Fe4+=O or Trp·+, during substrate (S) oxidation (to S• radical); and iii) CII reduction to RS during second substrate oxidation.
Despite its lower reduction potential, lignin oxidation by ApeLiP was not compromised. Transient-state kinetics with lignosulfonate substrates (Table 1) confirmed that ApeLiP can oxidize lignin from different origins with similar efficiency (k3app) values, unlike previously reported for P. chrysosporium LiPA [59]. Moreover, ApeLiP was still active after blocking the phenolic groups by acetylation of lignosulfonates, proving its capability to oxidize the major non-phenolic lignin moiety. Comparing the reduction of CII to RS enzyme, the rate-limiting step in lignin oxidation, the observed transient-state rate constants for ApeLiP were similar or slightly higher than those reported for P. eryngii VPL [27] and P. chrysosporium LiPA [59]. Moreover, in agreement with results using model compounds, we barely detected any activity on lignosulfonates with the W166A variant, definitively confirming the role of Trp166 oxidizing both the phenolic and non-phenolic units of lignin. Interestingly, the efficiency of ApeLiP oxidizing the acetylated softwood lignosulfonate was higher than those of P. chrysosporium LiP and P. eryngii VP acting on the same native lignosulfonate, a result that reveals this Agaricales LiP as the best biocatalyst oxidizing softwood lignin described to date.
|
Table 1. Transient-state kinetic parameters -KD3 (µM), k3 (s-1) and k3app (s-1·mM-1)- for rate-limiting CII reduction of ApeLiP, its W166A variant, P. eryngii VPL and P. chrysosporium LiPA (isoenzyme H8) by native and acetylated softwood and hardwood lignosulfonates.a |
|||||
|---|---|---|---|---|---|
|
Softwood lignin |
Hardwood lignin |
||||
|
Native |
Acetylated |
Native |
Acetylated |
||
|
ApeLiP |
KD3 |
nsb |
31±6.9 |
156±25 |
89±14 |
|
k3 |
ns |
26±1.7 |
163±38 |
56±4.6 |
|
|
k3app |
926±47 |
830±195 |
957±270 |
630±111 |
|
|
W166A |
KD3 |
- |
- |
- |
- |
|
k3 |
- |
- |
- |
- |
|
|
k3app |
0 |
0 |
0 |
0 |
|
|
P. eryngii VPLd |
KD3 |
143±19 |
24±1.9 |
14±1 |
21±2.5 |
|
k3 |
48±2 |
14±0.4 |
14±2 |
12±0.5 |
|
|
k3app |
340±30 |
599±31 |
990±80 |
592 ±52 |
|
|
P. chrysosporium LiPAe |
KD3 |
95±26 |
nac |
19±2 |
na |
|
k3 |
25±4 |
na |
14±0 |
na |
|
|
k3app |
263±83 |
na |
764±86 |
na |
|
|
aReactions in 0.1 M sodium tartrate, pH 3, at 25°C. bns, non-saturation kinetics. cna, not available. dfrom [23]. efrom [50]. Means and 95% confidence limits of replicate assays. |
|||||
Finally, using 2D-NMR, a considerable decay of softwood lignosulfonate was observed, and even more of hardwood lignosulfonate, resulting in strong decreases in the intensities of the lignin aromatic signals. In contrast, only slight modification in the proportion of lignin substructures with different inter-unit linkages was observed (Table 2). Interestingly, ApeLiP causes stronger modification of G lignin than P. chrysosporium LiPA in steady-state treatments under similar reaction conditions [59]. The above was not only evidenced by stronger lignin modification when using softwood lignosulfonate as ApeLiP substrate, but also by the preferential removal of G units during the treatment of hardwood lignin. The results suggest that ApeLiP could play a role in plant biomass degradation by A. pediades, and that LiPs with relevant ligninolytic capabilities, resulting from an evolutionary pathway different from those of Polyporales LiPs [29], exist in Agaricales.
|
Table 2. Semiquantitative HSQC-NMR analysis of lignosulfonates after 24-h treatment with ApeLiP including: a) Lignin composition (G and S units) and decay (parentheses); and b) Side-chain linkages as relative percentages and referred to 100 lignin units (parentheses) |
||||
|
Softwood lignosulfonate |
Hardwood lignosulfonate |
|||
|
Control |
ApeLiP |
Control |
ApeLiP |
|
|
a) Lignin composition and decaya |
||||
|
Guaiacyl units (G) |
126 (100) |
36 (71) |
12 (100) |
1 (92) |
|
Syringyl units (S) |
0 |
0 |
27 (100) |
5 (82) |
|
Total lignin |
126 (100) |
36 (71) |
39 (100) |
6 (85) |
|
Lignin S/G ratio |
0 |
0 |
2.2 |
4.7 |
|
b) Side-chain inter-unit linkagesb |
||||
|
β-O-4′ ethers (A) |
87 (33) |
90 (28) |
88 (31) |
89 (28) |
|
Phenylcoumarans (B) |
9 (4) |
6 (2) |
4 (1) |
4 (1) |
|
Resinols (C) |
4 (2) |
4 (1) |
8 (3) |
7 (2) |
|
Total |
100 (38) |
100 (31) |
100 (35) |
100 (32) |
|
aRelative intensities (DMSO signal= 100) of G and S signals (with decay percentages in parentheses); bPercentage (A+B+C= 100) of lignin substructures with different side-chain linkages (with values referred to total lignin units [G+S= 100] in parentheses) |
||||
5. Conclusions
Mushroom ligninolytic machineries have been understudied despite being potential sources of oxidoreductases with biotechnological potential. ApeLiP crystal structure, kinetic characterization, and the thorough study of its ligninolytic capability shows that this enzyme is able to oxidize both phenolic and non-phenolic lignin model compounds and real lignin, the latter in similar or higher extent than other well-known ligninolytic peroxidases. Therefore, not only wood-rotting Polyporales, but also Agaricales mushrooms, have enzymes with high relevance for both carbon recycling in nature and biotechnological modification of lignin.