The involvement of impaired alpha (α) cell function has been recognized as playing an essential role in several diseases, since hyperglucagonemia has been evidenced in both Type 1 and T2DM. This phenomenon has been attributed to intra-islet defects, like modifications in pancreatic α cell mass or dysfunction in glucagon’s secretion. Emerging evidence has shown that chronic hyperglycaemia provokes changes in the Langerhans’ islets cytoarchitecture, including α cell hyperplasia, pancreatic beta (β) cell dedifferentiation into glucagon-positive producing cells, and loss of paracrine and endocrine regulation due to β cell mass loss. Other abnormalities like α cell insulin resistance, sensor machinery dysfunction, or paradoxical ATP-sensitive potassium channels (KATP) opening have also been linked to glucagon hypersecretion.
- glucagon
- Langerhans’ islets
- type 2 diabetes
- hyperglycaemia
- hypoglycaemia
- α Cell
1. Introduction
2. Alpha Cell Physiology: From Secretion to Regulation
The human pancreas contains 1–2 million islets, each measuring 50–100 μm in diameter and containing ∼2000 cells on average. However, islet cells are only 2% of the overall pancreatic mass. It is notable that up to 65% of the human islet cells are α cells [8][10]. All islet cells originate from the endoderm. Its differentiation into each islet linage is mediated by the pancreatic and duodenal homeobox 1 (Pdx1) and neurogenin-3 (Ngn3) genes. The further evolution of α cells requires both aristaless-related homeobox (Arx) and forkhead box protein A2 (Fox-A2) action in addition to low expression levels of paired box 4 (Pax4). Other factors important for α cell differentiation include MAF BZIP Transcription Factor B (MafB), NK6 Homeobox (Nkx6.1; Nkx6.2), Pax6 [9][11], and RNA Paupar (PAX6 Upstream Antisense RNA). The latter is a novel long noncoding that has been shown to regulate α cell development through alternative splicing of Pax6 [10][12].
Glucagon is the primary α cell hormone product. It is derived from the proteolysis of the 160-aminoacid pre-pro-glucagon peptide coded by its gene located in 2q24.216. This gene is strongly expressed in α cells, the brain, and L-cells of the gut. Under normal conditions, α cells synthetize glucagon via proconvertase 2 post translational proteolysis, while L-cells produce glucagon-like peptide-1 (GLP-1) via proconvertase 1/3 pathway.
The classic model of glucagon secretion regulation is explained by glucose-medicated glucagon exocytosis. Anatomically, pancreatic islets are highly vascularized to ensure a rapid glucose and aminoacidic sensing. A glycaemic drop near to threshold stimulates glucagon release [11][17]. The cellular mechanism behind this glucose-dependent regulation of glucagon secretion is described in Figure 1.
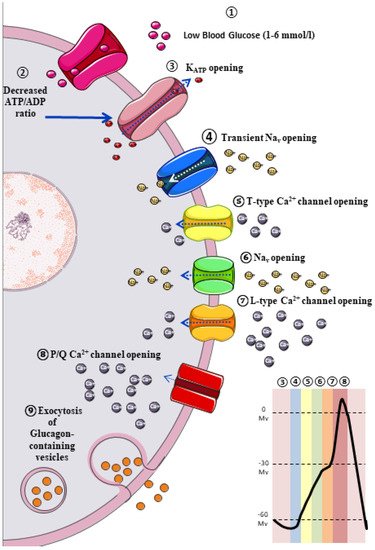
3. Extrinsic Model for the Regulation of Glucagon Secretion: Neurohormonal Mechanism
3.1. Paracrine Regulation
Omar-Hmeadi et al. observed a lack of inhibition in glucagon exocytosis by hyperglycaemia, somatostatin, or insulin in intact islets in α cells from T2DM cadaveric. Instead, hyperglycaemia inhibits α cell exocytosis, but not in the T2DM donor’s α cell or when paracrine inhibition by insulin or somatostatin is blocked. A reduced Surface expression of Somatostatin-receptor-2 in islet from T2DM donors suggests somatostatin resistance, and consequently, elevated glucagon in T2DM may reflect α cell insensitivity to paracrine inhibition during hyperglycaemia [12][16].
3.2. Autocrine Regulation
It is widely known that α cell membranes contain a large number of glucagon receptors (GR) [13][32]. Furthermore, a recent study reported that GR activation increases glucagon gene transcription via cAMP response element-binding (CREB) activation by the PKA-dependent pathway [14][40]. Moreover, several studies have reported that blocking GRs has been reported to improve glucose homeostasis [15][16][17][41,42,43], due to glucagon stimulates its own release acting on GR on α cells [18][44]. In fact, in a study conducted by Liu et al. on αTC1 cells (a pancreatic alpha cell line derived from an adenoma created in transgenic mice), immunofluorescent staining confirmed the presence of GRs on αTC1 cells. After 72 h of treatment with GRs antagonist (to block the effects of the endogenous glucagon), a 44% decrease in αTC1 cell proliferation was observed compared with the control group by counting the cells in the S phase. These results show that glucagon has direct trophic effects on α cells by an autocrine mechanism, and when the pancreatic α-cell number was decreased in db/db mice by a glucagon receptor antagonist, plasma glucagon levels were significantly decreased too [19][45].3.3. Juxtacrine Regulation
The juxtacrine mechanism is a common type of signalling between adjacent cells requiring direct contact between cells. Recently, a juxtacrine connection between α- and β-cells has been documented as another exciting way in glucagon secretion control since the confirmation of Eph/ephrin system between α- and β cells [20][21][22][51,52,53]. The Eph signalling system belongs to the superfamily of transmembrane Tyr kinase receptors. Today, it is well established that Eph allows short-distance cell-cell interaction by binding with their specific ligands (ephrin), primarily affecting cytoskeleton and leading to cell repulsion or adhesion in some circumstances. In this regard, the first studies in this field showed that many processes involving fast changes in cellular morphology were ephrin–Eph dependent [23][54]. More recently, other critical ephrin–Eph signalling-mediated processes have been identified and characterised, e.g., axon guidance, synaptic plasticity, cancer, and processes like juxtacrine hormones release control involving short-distance cell-cell communication. In addition, there is increasing evidence about its influence on cell differentiation, proliferation, and apoptosis regulation [24][55].3.4. Endocrine Regulation
Non-pancreatic hormones also have an active role in α cell regulation. For example, Bagger et al. [25][66] demonstrated that intravenous (IV) glucose administration did not reduce glucagon levels in subjects with T2DM; however, it was achieved with oral glucose administration, suggesting a potential role of intestine lining and incretins on α cell. In addition, GLP-1 and gastric inhibitory polypeptide (GIP) are hormones encoded by the GCG gene (the same encodes glucagon), secreted by enterocytes, with well-studied insulinotropic effects [26][67]. However, considering the difficulty of these experimental studies, its activity on α cells remains disputed [26][27][67,68]. The regulation of glucagon secretion relies on both intrinsic mechanisms in the α cell and extrinsic mechanisms in neighbouring and nearby cells since even autacoids like serotonin exhibit receptors in the α cell membrane inhibiting glucagon release. When α cells have a weak serotonergic tone, they lose their glucose-dependent inhibition of the glucagon-release, generating hyperglucagonemia under hyperglycaemic conditions.4. Hyperglucagonaemia and α Cell Dysfunction in Diabetes
Extensive emerging evidence has been published among the “bi-hormonal theory” in T2DM pathogenesis in which the coexistence of hyperglucagonemia and relative insulin deficiency increase gluconeogenesis and exacerbates peripheral IR [28][74], leading to overt T2DM development. Currently, there is some consensus regarding hyperglucagonemia origin. This is centred on two possible mechanisms: (1) a progressive loss in the regulatory mechanisms in the secretion patterns due to α cells functional alterations [29][30][75,76], or (2) modification in both the islet microarchitecture and cellularity [31][32][33][34][77,78,79,80].
4.1. Structural Alterations in Pancreatic Islet
Conclusive evidence has demonstrated a β cell mass reduction and a concomitant decrease in insulin secretion in subjects with long-standing T2DM [35][36][62,63] and a sensible fall in GABA and serotonin release, which are crucial paracrine regulators of glucagon secretion, as explained previously. However, post-mortem studies have reported an increased α cell mass in subjects with diabetes. Nonetheless, the evidence is not conclusive [36][63]. This finding can be related to a loss of α cell regulating factors or a compensatory mechanism secondary to β cells mass loss [36][63]. Nevertheless, multiple studies have reported that an elevation of Interleukin 6 (IL-6) circulating levels in T2DM adult mice is possibly linked with an expansion of α cell mass and hyperglucagonemia [37][64], suggesting that α cell proliferation in Type 1 Diabetes Mellitus (T1DM) is probably IL-6-dependent [38][81].