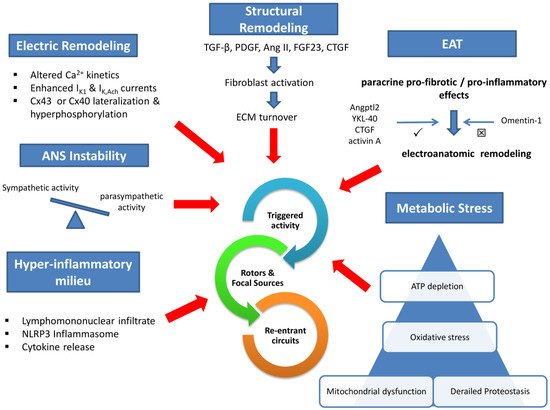
Electrical remodeling lies on impaired calcium handling, enhanced inwardly rectifying potassium currents, and gap junction perturbations. In addition, a wide array of profibrotic stimuli activates fibroblast to an increased extracellular matrix turnover via various intermediaries. Concomitant dysregulation of the autonomic nervous system and the humoral function of increased epicardial adipose tissue (EAT) are established mediators in the pathophysiology of AF. Local atrial lymphomononuclear cells infiltrate and increased inflammasome activity accelerate and perpetuate arrhythmia substrate. Finally, impaired intracellular protein metabolism, excessive oxidative stress, and mitochondrial dysfunction deplete atrial cardiomyocyte ATP and promote arrhythmogenesis.
1. Introduction
Cardiac senescence, largely attributed to aging, hypertension, obesity, as well as genetic predisposition, has been associated with atrial fibrillation (AF) genesis and progression
[1]. AF is expected to affect 6–16 million individuals in the United States, 14 million in Europe, and 72 million in Asia by 2050
[2], imposing a surge with economic and social implications for the public health care systems.
One out of three patients with AF will develop heart failure, and 20–30% of ischemic strokes are attributed to AF, increasing morbidity and mortality
[3].
Almost four centuries have elapsed since 1628 when William Harvey was probably the first to describe AF in animals
[4]. Nowadays, different ablation strategies have revolutionized AF treatment. However, a ‘’ceiling’’ for more durable long-term success (strictly defined as no occurrence of AF) seems to be reached. Success rates range between 65 and 78%, requiring an in-depth understanding of mechanistic links
[5].
2. Atrial Fibrillation Pathogenesis
2.1. Mechanistic Approach
Increased focal atrial triggered activity, mainly due to delayed afterdepolarizations (DADs) and micro-reentrant circuits are the main electrophysiological mechanisms in all types of AF (paroxysmal, persistent, and permanent)
[6].
In 1998, Haïssaguerre M et al. suggested that pulmonary vein (PV) ectopic activity is implicated in AF pathogenesis, paving the way for pivotal ablative therapeutic modalities such as pulmonary vein isolation (PVI)
[7]. Abnormal atrial repolarization (exaggerated beat-to-beat oscillations in action potential duration [APD]) and decreased atrial conduction are shown to mitigate re-entry in patients with AF
[8][9][8,9].
A frequency-domain approach, utilized to explain AF initiation and maintenance, underscores that AF mechanistic links might be less chaotic than originally thought. In particular, focal impulse and rotor modulation (FIRM) mapping has already achieved a ‘’panoramic’’ bi-atrial view and suggests that a small number of stable high-frequency re-entrant sources (rotors) perpetuate AF fibrillatory waves. This spatiotemporally ordered AF substrate was successfully targeted via FIRM-guided ablation with salutary long-term effects in CONFIRM and RADAR AF trials
[10][11][10,11].
2.2. Molecular Pathophysiology
2.2.1. Ionic Perturbations
The study of electric remodeling in human atria focuses, mainly, on altered calcium kinetics, impaired inwardly rectifying potassium currents, and gap junction changes.
Abnormal intracellular calcium (Ca
2+) handling is critical in triggering DADs and thus increased atrial ectopic activity. In human AF models, enhanced spontaneous sarcoplasmic reticulum (SR) Ca
2+ release has been attributed to ryanodine receptor (RyR2) dysregulation
[12][13][14,15], Ca
2+/calmodulin-dependent protein kinase-II (CaMKII) hyperactivity
[14][15][16][16,17,18], or SPEG (striated muscle preferentially expressed protein kinase), a regulator of RyR2 phosphorylation and downregulation
[17][19].
L-type Ca
[2] current (I
Ca,L) attenuation leads to atrial APD shortening and seems to be implicated in AF maintenance
[18][19][20,21]. An effect is at least partially driven by microRNA-21 and microRNA-328 in humans
[20][21][22,23]. Early re-activation of I
Ca,L current can also lead to early afterdepolarizations (EADs) and AF initiation
[22][24]. Interestingly, impaired calcium homeostasis in human cardiomyocytes leads to inositol-trisphosphate-receptor (IP3R)/CAMKII signaling, which in turn decrease I
Ca,L density
[23][25].
Inwardly rectifying potassium (Kir) current (I
K1) as well as acetylcholine-activated potassium current (I
K,Ach) are enhanced in AF patients and shorten atrial APD
[24][25][26,27]. MicroRNA-26 and microRNA-1 downregulation leads to increased Kir2.1 protein expression and establishes atrial re-entry via I
K1 current activation
[26][27][28,29]. Both I
K1 and I
K,Ach currents are critically involved in maintaining a left-to-right dominant frequency gradient in paroxysmal AF (PAF) subjects and explain AF drivers formation (rotors and focal impulses)
[28][30].
Cardiac connexins create gap junctions, facilitating cell-to-cell electrical and molecular signaling
[29][31]. Ultra-structural changes of atrial connexins are noticed in human AF experiments. Connexin-43 (Cx43) dysregulation is present in the atria of AF patients
[30][32] and seems to be regulated through interplay between microRNA-613 and long noncoding RNA HOTAIR (HOX transcript antisense RNA)
[31][33]. Connexin-40 (Cx40) is mainly found in the atrial myocardium and is associated with AF development, as shown in various genetic analyses
[32][33][34][34,35,36]. Increased lateralization and hyperphosphorylation of either Cx43 or Cx40 are implicated in human AF pathophysiology
[35][36][37][37,38,39].
2.2.2. Structural Changes
Atrial fibrosis is the result of increased fibroblast activity with heterogeneous patchy areas of collagen type I depositions. This favors longer AF periods, LA enlargement, distortion of intercellular electrical coupling, and perpetuation of AF fibrillatory waves
[38][40].
Of note, delicate 3D human atria models suggested that fibrosis reduces atrial conduction velocity and stabilizes rotors and re-entrant circuits
[39][41]. A study including patients with persistent AF, who underwent LA tissue characterization with MRI scans and concomitant high-density mapping of the LA, demonstrated increased AF rotor activity in areas of relatively low and patchy late-gadolinium enhancement (LGE)
[40][42]. Bioinformatics’ analysis has recently revealed that LA–PV junction demonstrates distinct gene expression differences in AF patients as compared to sinus rhythm (SR) controls, favoring extracellular matrix (ECM) synthesis and chemokine up-regulation
[41][43].
TGF-β signaling is strongly associated with these structural changes
[42][44]. Various microRNAs and long noncoding RNAs seem to regulate cardiac fibroblast profibrogenic activity
[43][44][45][46][47][45,46,47,48,49]. A recently discovered crosstalk between Slit2-Robo1 and TGF-β1/Smad pathways promises potential therapeutic targets against atrial fibrosis
[48][50].
Furthermore, platelet-derived TGF-β secretion stimulates fibroblast proliferation, setting a vicious cycle of atrial fibrosis
[49][51]. Platelet-derived growth factor (PDGF) also leads to increased cardiac fibroblast activity
[50][52] and eventually to atrial fibrosis
[51][53].
Mitogen-activated protein kinase 1 (MAPK1) overexpression, evident in cardiac fibroblast from AF patients, stimulates collagen deposition. An effect that can be mitigated by microRNA-450a-2-3p
[52][54].
Angiotensin II (Ang II) is also known to induce critical ECM changes (increased collagen deposition and metalloproteinase activity) via JAK/STAT3 molecular pathway
[53][55]. Ang II receptor type 1 (AT1) is up-regulated in the LA of subjects suffering AF
[54][56]. Inhibition of AT1 restores intracellular calcium homeostasis and prevents arrhythmogenesis
[55][57]. In addition, fibroblast growth factor 23 (FGF23) is involved in atrial fibrogenesis via increased oxidative stress and STAT3/SMAD3 signaling
[56][58].
Connective tissue growth factor (CTGF) levels in human atrial fibroblasts and epicardial adipose tissue (EAT) are positively correlated with atrial fibrosis and AF arrhythmogenesis
[57][58][59,60]. MicroRNA-132 and Ang II regulate CTGF levels in human atria
[59][60][61,62].
Finally, atrial tissue calcitonin levels are inversely correlated with atrial arrhythmogenesis. A recent study suggested that calcitonin halts cardiac fibroblast overactivity and prevents ECM turnover
[61][63].
Apart from generating a fibrotic substrate, cardiac fibroblasts affect cell-to-cell electrical coupling and exhibit altered electrophysiological properties in humans suffering from AF compared to SR controls
[62][64]. This finding necessitates further evaluation, especially in the light of mechanistic data suggesting that fibroblast proliferation leads to complex fractionated atrial electrograms (CFAEs) genesis
[63][65] and action potential propagation block in pulmonary veins
[64][66].
2.2.3. Epicardial Adipose Tissue (EAT) and Autonomic Nervous System (ANS)
From a mechanistic point of view, the EAT in patients with persistent AF (PeAF) seems to be related to rotors capable of maintaining AF
[65][67] and positively associated with low voltage areas, reduced conduction velocity, and CFAE
[66][67][68,69].
EAT is a known cause of electrophysiological changes, such as heterogeneous atrial conduction slowing. These alterations have been attributed to Cx40 lateralization, excessive fibrosis, and heterogeneous adipose infiltration of the affected atria
[68][70].
Interestingly, a unique molecular footprint has recently been shown in EAT from AF subjects. In particular, EAT derived extracellular vesicles (EVs) exert profibrotic/proinflammatory effects on the neighboring atrial tissue, promoting arrhythmogenesis
[69][71]. In addition, EAT expansion seems to be positively regulated through increased atrial natriuretic peptide (ANP) levels
[70][72].
EAT is metabolically active, and paracrine secretion of inflammatory mediators (IL-1β, among others)
[71][72][73,74] is associated with atrial fibrillation in humans
[73][75].
EAT-mediated atrial fibrosis has been linked to the PeAF subtype, and CD8
+ lymphocyte infiltrates are seemingly involved
[74][76]. Angiopoietin-like protein 2 (Angptl2), YKL-40, CTGF, activin A (TGF-β superfamily) upregulation and Omentin-1 downregulation in human EAT are also implicated in atrial fibrosis and AF development
[58][75][76][77][78][60,77,78,79,80].
Intrinsic cardiac ANS is organized in a network of ganglionated plexi (GP), which are accommodated in EAT, mainly around PVs
[79][81].
Various methods of cardiac ANS assessment (heart rate variability,
[80][81][82,83] skin sympathetic nerve activity
[82][84], metaiodobenzylguanidine (MIBG) scintigraphy
[83][85]) imply that ANS instability is implicated in human AF pathogenesis.
The clinical impact of ANS modulation (GP ablation in addition to PVI in patients with PAF
[84][86], chemical; botulinum toxin
[85][86][87,88] or calcium chloride
[87][89]; autonomic denervation in cardiac surgery patients, transcutaneous vagal nerve stimulation
[88][90]) in managing AF further strengthens the role of autonomic remodeling in AF pathophysiology, and it is further discussed in
Section 3: Therapeutic Perspectives.
2.2.4. The Role of Inflammation
Local inflammation is apparent in AF pathophysiology since human LA tissue examination has revealed an infiltrate of varying immune cells (neutrophils, proinflammatory CD68
+ macrophages, CD8
+ and CD3
+ lymphocytes) in AF subjects
[89][90][91][92][91,92,93,94].
IL-6 secretion seems to be a critical mediator in suppressing regulatory T cell function and triggering atrial fibrosis
[93][95]. In addition, macrophage migration inhibitory factor (MIF) release, an early mediator in inflammation cascade, was previously shown to suppress I
(Ca,L) current and is also associated with AF genesis
[94][96].
Serum levels of Interleukin-2 soluble receptor and TNF-α soluble receptor are among the stronger predictors of new-onset AF, as assessed via machine learning algorithms in Multi-Ethnic Study of Atherosclerosis (MESA)
[95][97]. This observation is in accordance with other indices of systematic inflammation (TNF-α, hs-CRP, IL-6, IL-8, and IL-18)
[96][97][98][99][98,99,100,101], all of which are up-regulated in the serum of patients with AF. These findings suggest the interplay of an atrial-specific and systematic hyperinflammatory state in AF subjects. Additionally, elevated baseline hs-CRP levels independently predicted arrhythmia recurrence post-ablation and are positively associated with low LA voltage areas, rotors, and non-PV ectopic foci
[100][102].
NLRP3 (NACHT, LRR, and PYD domain-containing protein 3) inflammasome activity is also enhanced in atrial cardiomyocytes from AF patients and brings about electroanatomic remodeling
[101][103]. Post-operative AF (POAF) patients were shown to express a higher level of the activated inflammasome in their atrial tissue, an observation linked to enhanced spontaneous SR Ca
2+ release and DADs formation
[102][104].
From a clinical perspective, recent evidence suggests that immunomodulatory agents, such as corticosteroids and colchicine, have a preventive role in POAF development
[103][104][105][106][105,106,107,108] and support the fundamental role of inflammatory pathways in managing AF.
2.2.5. The Role of Proteostasis, Oxidative Stress, and Mitochondrial Bioenergetics
The role of metabolic stress is increasingly recognized in AF pathophysiology, and it is discussed below in view of impaired protein cycling, oxidative stress, and mitochondrial dysfunction.
Proteostasis is defined as the balance between protein synthesis, folding, and degradation
[107][109]. Impaired protein homeostasis is observed in human cellular aging as well as in cardiac diseases
[108][110]. Derailed proteostasis exhibited through heat shock proteins (HSPs) up-regulation, calpain hyperactivity, and autophagosome formation is involved in AF genesis.
HSPs are produced as a response to cellular stress and stabilize other intracellular proteins. HSP27 was previously shown to be up-regulated in the atria of PAF patients and attenuates stress-induced structural changes (myolysis)
[109][111]. In addition, low baseline HSP27 is associated with low LA voltage areas, non-PV foci, and decreased arrhythmia free intervals in patients undergoing ablation for PAF
[110][112]. A more recent study suggests that post-ablation rise in serum HSP27 levels are predictive of arrhythmia recurrence, while baseline levels of different HSPs are of no clinical significance, thus creating a need for further research
[111][113].
Atrial tissue from PeAF patients demonstrates increased macroautophagy (a process of autophagosome formation and eventually lysosomal degradation of damaged proteins), which is linked to reduced I
(Ca,L) current and atrial APD shortening in animal studies
[112][114].
Calpain I (a non-lysosomal proteolytic enzyme) activity is enhanced in atrial myocytes of both PAF and PeAF patients and has been linked with APD shortening
[113][115]. Histone deacetylase 6 (HDAC6) hyperactivity, evident in human AF atria, disrupts cytoskeleton (microtubules) and culminates in increased α-tubulin degradation by calpains
[114][116]. Recently, HDAC6 up-regulation was proven capable of triggering atrial fibrosis and Cx lateralization in a rat AF model
[115][117].
In PeAF patients, increased markers of DNA damage were positively associated with poly(ADP-ribose) polymerase (PADP) levels and hint an energy-deficient state. In particular, the physiologic cellular process of DNA repair sometimes leads to exaggerated PADP activity and nicotinamide adenine dinucleotide (NAD
+) depletion, which in turn confers oxidative stress and progressive ATP decline
[116][118].
Additionally, increased production of reactive oxygen species (ROS) is implicated in human AF via both local (atrial cardiomyocyte) and systematic (serum) nicotinamide-adenine dinucleotide phosphate oxidase (NOX) activity
[117][118][119,120]. Low levels of DNA oxidative stress markers in serum or urine from AF patients have been associated with prolonged arrhythmia-free survival
[119][120][121,122].
Finally, mitochondrial energy production is critically affected in AF patients since reduced oxidative phosphorylation and increased mitochondrial fragmentation lead to ATP depletion
[121][122][123,124]. POAF patients are also known to exhibit impaired oxidative phosphorylation capacity pre-operatively
[123][125].
Long noncoding RNAs might be involved in mitochondrial bioenergetics, regulating ATP synthase and CYP450 enzymes
[124][126]. Mitochondrial dysfunction is postulated to induce electrical remodeling via oxidative dysfunction of RyR2
[125][127].
Adenosine monophosphate-regulated protein kinase (AMPK) activity demonstrates a compensatory increase as a response to AF-induced metabolic stress, restoring calcium homeostasis
[126][128], and it is suggested to be a novel therapeutic target.
Evidently, AF pathogenesis involves overlapping cellular and molecular perturbations that hinder us from distinguishing the cause from the effect (see
Fugure 1Appendix A). Since gauging the critical importance of any single mechanism in different clinical AF subtypes is both impractical and unsettled, many therapeutic strategies target multiple mechanisms and seem promising, as discussed in the following section.
Figure 1. Central Illustration Legend: Six overlapping and interlinked (not shown) pathways are implicated in atrial fibrillation pathophysiology: electric remodeling, structural remodeling, autonomic instability, hyperinflammatory milieu, metabolic stress, and epicardial adipose tissue paracrine effects. The common endpoint is the initiation and maintenance of arrhythmic events. IK1—inwardly rectifying potassium current; IK,Ach—acetylcholine-activated potassium current; ANS—autonomic nervous system; Cx—connexin; TGF-β—tissue growth factor β; PDGF—platelet-derived growth factor; AngII—angiotensin II; FGF23—fibroblast growth factor 23; CTGF—connective tissue growth factor; ECM—extracellular matrix; EAT—epicardial adipose tissue; Angptl2—angiopoietin-like protein 2.