Hyperuricemia is significantly associated with the development and severity of the metabolic syndrome. The increased urate transporter 1 (URAT1) and glucose transporter 9 (GLUT9) expression, and glycolytic disturbances due to insulin resistance may be associated with the development of hyperuricemia in metabolic syndrome. Hyperuricemia was previously thought to be simply the cause of gout and gouty arthritis.
- cardiovascular diseases
- chronic kidney disease
- hyperuricemia
- uricosuric
- xanthin oxidase
1. Introduction
Uric acid (UA) is synthesized mainly in the liver, intestines, and vascular endothelium as the end product of an exogenous purine from food (100–200 mg/day), and endogenously (500–600 mg/day) from damaged, dying, and dead cells, whereby nucleic acids, adenine and guanine, are degraded into UA [1]. There are two types of nucleotide production pathways: a de novo synthesis which newly creates purines and a salvage pathway that reuses purines ( Figure 1 ). Such nucleotides are metabolized to xanthine, which is finally converted to UA by the action of xanthine oxidase (XO). Approximately daily 700 mg of UA is produced by such processes. The kidney plays a dominant role in UA excretion, and the kidney excretes approximately 70% of daily produced UA [2]. The remaining 30% of UA is excreted from the intestine [3]. Usually, the UA pool size of an adult male is about 1200 mg [4]. The UA production is balanced by the excretion of UA into urine (500 mg) and intestine (200 mg). When UA production exceeds UA excretion, hyperuricemia, which has been defined as serum UA concentration > 7.0 mg/dL [5], occurs.
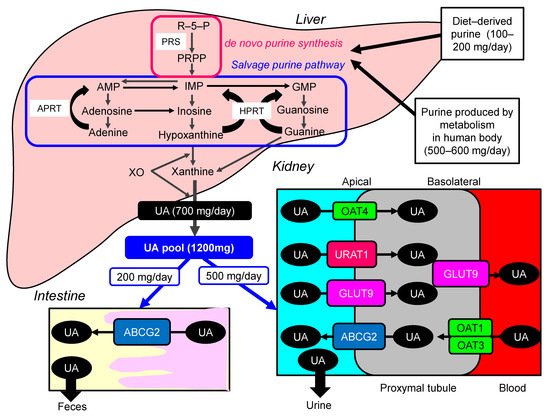
Hyperuricemia is induced by UA over-production due to acquired factors such as high purine diet, fructose ingestion, alcohol intake, myeloproliferative disorders, and also rare genetic causes such as hypoxanthine-guanine phosphor-ribosyl-transferase (HPRT) deficiency and phosphor-ribosyl-pyrophosphate (PPRP) synthetase (PRS) hyperactivity [6]. Renal excretion of UA is the major regulator of serum UA concentration [7], [8]. In humans, reabsorption of UA into the blood plays a crucial role to regulate serum UA. The UA exchange is mediated by various molecules expressed in renal proximal tubule [9]. Such molecules include glucose transporter 9 (GLUT9) [10], urate transporter 1 (URAT1) [11], and human ATP-binding cassette, subfamily G, 2 (ABCG2) [12], organic anion transporter (OAT)1, 3, and 4 [6]. UA enters the cell in exchange for monocarboxylate via apical URAT1 and for dicarboxylate via apical OAT4 [6]. OAT1 and OAT3, on the basolateral membrane of epithelial cells, transport UA from the renal interstitial into renal proximal tubule epithelial cells [13]. Renal UA reabsorption is mainly mediated by URAT1 and GLUT9 [11][14][15][11,14,15]. URAT1 is found in the apical membrane of proximal tubule epithelial cells [6]. Apical GLUT9 plays a significant role in UA reabsorption, the reabsorbed UA exiting the cell through basolateral GLUT9 [6]. In addition, ABCG2 has been identified as a high-capacity UA exporter that mediates renal and/or extra-renal UA excretion [16]. ABCG2 is now known to be involved as well in UA excretion into the intestine [16].
Hyperuricemia has been previously classified into the “UA overproduction type”, “UA underexcretion type” and “combined type”, and approximately 10%, 60%, and 30% of hyperuricemic patients have been classified into each type, respectively [5][17][5,17]. Abcg2-knockout mice showed increased serum UA and renal UA excretion, and decreased intestinal UA excretion [18], indicating that a significance of decreased extra-renal UA excretion caused by ABCG2 dysfunction for hyperuricemia. At present, hyperuricemia is classified into “renal UA overload type” (“UA extra-renal underexcretion type” and “UA overproduction type”), “UA underexcretion type” and “combined type” [5].
Hyperuricemia was previously thought to be simply the cause of gout and gouty arthritis. Further, the hyperuricemia observed in patients with renal diseases was considered to be caused by UA underexcretion due to renal failure since the kidney excretes approximately 70% of daily produced UA [2]. Therefore, hyperuricemia was not considered as an aggressive treatment target. Here, we show that hyperuricemia itself is associated with the development and severity of metabolic syndrome, cardiovascular diseases (CVD) and chronic kidney disease (CKD), and possible beneficial effects of the treatment for hyperuricemia on CVD and CKD.
2. Hyperuricemia and Metabolic Syndrome
Hyperuricemia is significantly associated with the development and severity of metabolic syndrome. Meta-analysis showed that higher serum UA levels led to an increased risk of metabolic syndrome regardless of the study characteristics, and were consistent with a linear dose-response relationship [19]. Choi HK, et al. determined the prevalence of the metabolic syndrome at different serum UA levels by using data from 8669 participants aged 20 years and more in The Third National Health and Nutrition Examination Survey (1988–1994) [20]. They found that the prevalence of metabolic syndrome increases substantially with increasing serum UA levels. A nested case-cohort study of 431 patients with 220 cases demonstrating new vascular events during follow-up, originating from the Second Manifestations of Arterial Disease study, showed that serum UA levels were higher in patients with the metabolic syndrome than in patients without. Serum UA concentrations increased with the number of components of the metabolic syndrome adjusted for age, sex, creatinine clearance, and alcohol and diuretic use [21].
Takahashi, S, et al. evaluated the effect of accumulation of intraabdominal visceral fat on the metabolism of UA in 50 healthy male subjects [22]. Multivariate analyses showed that the size of the visceral fat area was the strongest contributor to an elevated serum UA and a decrease in UA clearance. Magnitude of insulin resistance and serum UA concentration were significantly related (r = 0.69; p < 0.001), and insulin resistance was also inversely related to urinary UA clearance (r = −0.49 ; p < 0.002), and urinary UA clearance was inversely related to serum UA concentration (r = −0.61; p < 0.001) [23]. Insulin resistance due to visceral fat accumulation may increase serum UA by decreasing renal UA clearance in patients with the metabolic syndrome.
Possible molecular mechanisms for the development of hyperuricemia in the metabolic syndrome were shown in Figure 2 . Over-intake of purine may be involved in the development of hyperuricemia in metabolic syndrome, because overeating and lack of physical activity are the main causes of metabolic syndrome. The increased protein level of URAT1 was observed in obesity/metabolic syndrome model mice [24][31]. Upon high-purine load, insulin resistance enhances UA reabsorption as manifested by up-regulated URAT1 expression and reduces UA excretion in the Otsuka-Long-Evans-Tokushima Fatty rats [25][32].
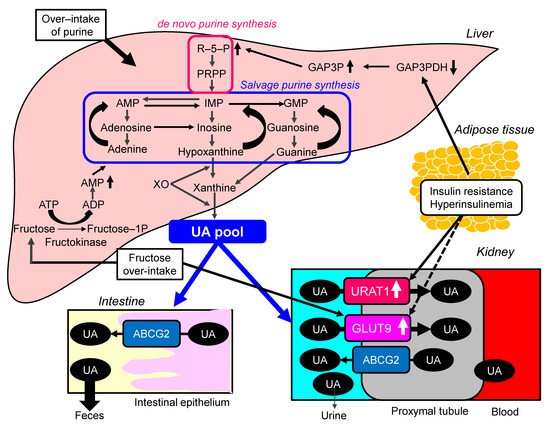
The meta-analysis of 29 diet-intervention trials showed that short-term fructose consumption promotes the development of hepatic insulin resistance in non-diabetic adults [26][34]. Another meta-analysis including 3102 articles suggested that fructose consumption from industrialized foods has significant effects on most components of metabolic syndrome [27][35]. The risk of hyperuricemia and gout was also positively correlated with the intake of fructose (OR, 2.14; 95%CI, 1.65 to 2.78) [28][36]. In the meta-analysis of prospective cohort studies, fructose consumption was associated with an increase in the risk of gout (RR, 1.62; 95%CI, 1.28 to 2.03; p < 0.0001) when comparing the highest and lowest quantiles of fructose consumption [29][37]. Fructose intake is significantly associated with the development of both hyperuricemia and metabolic syndrome.
3. Hyperuricemia and CVD
The meta-analysis including 15 studies showed that carotid intima-media thickness (CIMT) in the high UA group was significantly higher than that in the control group [standardized mean difference (SMD), 0.53; 95%CI, 0.38 to 0.68), and the difference was significant ( p < 0.00001) [30][42]. Subgroup analysis by disease status illustrated a positive relationship between serum UA levels and CIMT in healthy people and people with diseases. In the meta-analysis and systematic review to explore the modifiable risk factors for carotid atherosclerosis, hyperuricemia was significantly associated with the presence of carotid plaque, and could elevate the risk of atherosclerosis by at least 50% [31][43].
In the meta-analysis including 29 prospective cohort studies, hyperuricemia was associated with increased risk of CHD morbidity (adjusted RR, 1.13; 95%CI, 1.05 to 1.21) and mortality (adjusted RR, 1.27; 95%CI, 1.16 to 1.39) [32][44]. For each increase of 1 mg/dL in UA level, the pooled multivariate RR of CHD mortality was 1.13 (95%CI, 1.06 to 1.20). Another meta-analysis including 14 studies involving 341, hyperuricemia was associated with an increased risk of CHD mortality (RR, 1.14; 95 %CI, 1.06 to 1.23) [33][45]. For each increase of 1 mg/dL of serum UA, CHD mortality increased by 20%. According to the gender subgroup analyses, hyperuricemia increased the risk of CHD mortality in women (RR, 1.47; 95 %CI, 1.21 to 1.73) compared to men (RR, 1.10; 95 %CI, 1.00 to 1.19). In a systematic review and meta-analysis using 26 studies, hyperuricemia was associated with an increased risk of CHD incidence (unadjusted RR, 1.34; 95%CI, 1.19 to 1.49) and mortality (unadjusted RR, 1.46; 95%CI, 1.20 to 1.73) [34][46]. When adjusted for potential confounding, the pooled RR was 1.09 (95%CI, 1.03 to 1.16) for CHD incidence and 1.16 (95%CI, 1.01 to 1.30) for CHD mortality. For each increase of 1 mg/dL in UA level, the pooled multivariate RR for CHD mortality was 1.12 (95%CI, 1.05 to 1.19). Subgroup analyses showed no significant association between hyperuricemia and CHD incidence/mortality in men, but an increased risk for CHD mortality in women (RR, 1.67; 95%CI, 1.30 to 2.04).
The meta-analysis of 16 studies showed that hyperuricemia was associated with a significantly higher risk of stroke incidence (RR, 1.41; 95%CI, 1.05 to 1.76) and mortality (RR, 1.36; 95%CI, 1.03 to 1.69) [35][47]. Subgroup analyses of studies adjusting for known risk factors such as age, hypertension, diabetes mellitus, and cholesterol still showed that hyperuricemia was significantly associated with stroke incidence (RR, 1.47; 95%CI, 1.19 to 1.76) and mortality (RR, 1.26; 95%CI, 1.12 to 1.39). Another meta-analysis including 15 prospective studies indicated that the presence of hyperuricemia was associated with a significantly greater risk of stroke incidence (RR, 1.22; 95%CI, 1.02 to 1.46) and mortality (RR, 1.33; 95%CI, 1.24 to 1.43) [36][48]. In addition, the pooled estimate of multivariate RRs of stroke incidence and mortality were 1.08 (95%CI: 0.85 to 1.38); 1.26 (95%CI: 1.14 to 1.40) among men and 1.25 (95%CI: 1.04 to 1.46); 1.41 (95%CI: 1.31 to 1.52) among women respectively.
Oxidative stress is a well-known component of atherosclerotic pathogenesis, occurring in parallel with activation of pro-inflammatory signaling pathways and expression of cytokines/chemokines [37][51]. XO uses molecular oxygen as electron acceptor to generate hydrogen peroxide and superoxide anions [38][52]. XO is normally present in the endothelial cells and in blood, and its levels in atherosclerotic plaques was found to be increased [39][53]. Several studies highlighted the possible involvement of XO in atherosclerosis development. It was shown that atherogenesis in apo-E knockout mice could be reduced by XO inhibitors [40][54]. Further, the inhibition of XO reduced the endothelial dysfunction in heavy smokers [41][55]. XO stimulates the expression of scavenger receptors in macrophages and vascular smooth muscle cells.
4. Hyperuricemia and Renal Dysfunction
In the meta-analysis including 15 cohorts with a total of 99,205 individuals and 3492 incident CKD, the RR of CKD was 1.22 (95%CI, 1.16 to 1.28) per 1 mg/dL serum UA level increment [42][59]. The observed positive association was more pronounced among group with a mean age < 60 years (RR, 1.26; 95%CI, 1.21 to 1.31). However, no association was observed among studies with a mean age ≥ 60 years (RR, 1.04; 95%CI, 0.96 to 1.13). This mean age-related difference in the association between serum UA levels and CKD was significant ( p = 0.004). A systematic review and meta-analysis including 13 studies containing 190,718 participants showed a significant positive association between elevated serum UA levels and the new-onset CKD at follow-up (OR, 1.15; 95%CI, 1.05 to 1.25) [43][60]. Hyperuricemia was found be an independent predictor for the development of newly diagnosed CKD in non-CKD patients (OR, 2.35; 95%CI, 1.59 to 3.46). This association increased with increasing length of follow-up. No significant differences were found for risk estimates of the associations between elevated serum UA levels and developing CKD between males and females.
UA is known to induce hypertension through its effects on endothelial function and impaired production of nitric oxide [44][61]. Hypertension can be the initial trigger leading to subclinical renal damages [45][62]. In animal models, hyperuricemia caused hypertension through activation of both vasoactive and inflammatory processes that have multiple effects that include sodium retention and vascular constriction [46][63]. Histologic analyses showed the lesions that were similar to those seen in hypertension, with the presence of arteriolosclerosis and tubule-interstitial injury [47][64]. Serum UA levels were significantly correlated with vascular resistance at the afferent, but also efferent, arteriole, suggesting that hyperuricemia may be harmfully associated to glomerular perfusion [48][65]. Emerging evidence suggests a significant contribution of activation of the RAS by hyperuricemia to the development of CKD [49][66]. Renal vasoconstriction and reduced renal plasma flow can be induced by activation of RAS. Further, UA may increase oxidative stress, leading to mitochondrial dysfunction, over-secretion of pro-inflammatory cytokines, and proliferation of vascular smooth muscle cells. UA crystals can cause tubular damage through inflammation mediated by direct physical mechanisms.