Src, originally identified as an oncogene, is a membrane-anchored tyrosine kinase (TK) and the Src family kinase (SFK) prototype. SFKs regulate the signalling induced by a wide range of cell surface receptors leading to epithelial cell growth and adhesion. In the intestine, the SFK members Src, Fyn and Yes regulate epithelial cell proliferation and migration during tissue regeneration and transformation, thus implicating conserved and specific functions. In patients with colorectal cancer (CRC), SFK activity is a marker of poor clinical prognosis and a potent driver of metastasis formation. These tumorigenic activities are linked to SFK capacity to promote the dissemination and tumour-initiating capacities of epithelial tumour cells. However, it is unclear how SFKs promote colon tumour formation and metastatic progression because SFK-encoding genes are unfrequently mutated in human cancer. Here, we review recent findings on SFK signalling during intestinal homeostasis, regeneration and tumorigenesis, and discuss about therapeutic strategies to efficiently target Src signalling in CRC
- Src family kinases
- intestinal homeostasis
- cell signalling
- colon cancer
- cancer therapy
1. Introduction of SFKs
- Introduction of SFKs
Src, originally identified as an oncogene, is a membrane-anchored cytoplasmic TK that mediates signalling induced by a wide range of cell surface receptors [1][2]. Notably, Src is a master regulator of cell growth and migration induced by extracellular cues. Src is also the prototype of SFK that includes eight members (Src, Fyn, Yes, Lck, Fgr, Hck, Blk and Lyn) of which three (Src, Fyn and Yes) are widely expressed [3][2]. Src shares with the other SFKs a common modular structure formed by the membrane-anchoring SH4 region through lipid attachment (i.e., myristoyl), followed by an intrinsically disordered region named the unique domain (UD), and the SH3, SH2 and kinase domain [3][2]. SH4 contains also a palmitoylation site for membrane anchoring, except in Src and Blk. The kinase domain is bordered by two short regulatory sequences, named the linker and the tail, involved in the tight regulation of the kinase activity to prevent aberrant protein tyrosine phosphorylation. Crystallography studies revealed that intramolecular interactions are part of the mechanisms that control SFK catalytic activity [4]. Notably, phosphorylation of a conserved tyrosine in the tail (Tyr 530 in the human Src sequence) promotes SH2 intramolecular interactions that, combined with the SH3-linker interaction, stabilise the enzyme in an autoinhibited conformation. This Src phospho-regulatory mechanism is conserved in all SFKs and is mediated by the cytoplasmic TKs C-terminal Src Kinase (CSK) and CSK homologous kinase [5]. Disruption of any of the SH2 or SH3-mediated protein interactions or tyrosine phosphatase activity leads to the kinase active conformation. Catalytic de-repression enables SFK autophosphorylation of the activation loop (Tyr419 in the human Src sequence) that further supports the kinase active state [4]. In agreement with this model, stabilization of the enzyme in a de-repressed conformation by somatic mutation or protein association results in constitutive SFK activity that can lead to oncogenic properties [6].
Recent findings revealed additional unsuspected Src regulatory mechanisms involving its UD. This unstructured region of about 70–80 amino acids is conserved in vertebrates and is unique among the different SFKs (e.g., different sequences). Although Src UD function remained mysterious until recently, NMR analyses revealed that this region has a compact, yet highly dynamic structure, described as an intramolecular fuzzy complex [7]. NMR-guided mutations that affect UD-SH3 interactions revealed an essential role for this fuzzy complex in Src signalling leading to CRC cell migration [8]. Moreover, Src can dimerise through involvement of the UD in the binding to a hydrophobic pocket within the kinase domain of the dimeric partner [9]. A biophysical study showed that the Src SH4 domain has dimerisation capacity on its own, suggesting a complex mechanism underlying Src dimerisation [10]. Importantly, Src dimerisation may define a novel regulatory mechanism because it substantially enhances Src autophosphorylation and phosphorylation of selected substrates [9]. Interestingly, SFKs share well-conserved sequence features involving aromatic residues in their UDs, suggesting a similar UD-dependent regulatory region in the other SFKs [7].
Finally, emerging evidence supports the existence of an additional regulatory mechanism through Src myristoylation, as described for the cytoplasmic TK Abelson (ABL) where a myristoyl binding pocket in the kinase domain maintains ABL in an inactive state [11]. Structural analyses suggested, but did not confirm yet, the presence of a similar binding pocket in Src [12]. Nevertheless, Moasser et al. reported that Src dimerisation also involves the interaction of the myristoylated N-terminal region with the kinase domain pocket in trans [9]. Surprisingly, Pons et al. discovered an additional myristoyl binding site in Src-SH3, that contributes to Src membrane anchoring [13]. Interestingly this interaction is modulated by the fuzzy complex contained in the UD, suggesting a mechanism linking Src activation and membrane anchoring. Therefore, we predict an important role for the SH4 and the UD in controlling the Src topology at the membrane or the local microenvironment for substrate selection and signalling. Whether this myristoylation-switch mechanism is conserved in other SFKs is currently unknown. Overall, these recent findings uncover a much higher complexity of SFK regulation than previously expected, which may have important implications on the SFK’s oncogenic functions.
2. SFKs in Intestinal Homeostasis and Regeneration
- SFKs in Intestinal Homeostasis and Regeneration
Genetic analyses in animals established essential physiological roles for SFKs [14]. For instance, constitutive SFK gene knock-out experiments in mice revealed an important function for Src in early development, at least partially shared with Fyn and Yes. Specifically, Src-deficient mice die early after birth because of defects in bones where Src is normally highly expressed. Conversely, combined Src, Fyn and Yes gene inactivation leads to mouse embryonic lethality [14]. Consistent with this, disruption of the SFK negative regulator Csk leads to embryonic lethality with excessive tissue SFK activity, indicating that SFK regulation is essential for mouse development [14]. However, other genetic analyses revealed that Src, but not Fyn, is partly epistatic to the Csk gene, consistent with SFK partially redundant functions during development [13]. Then, tissue-specific gene manipulation studies showed important roles for SFKs in epithelial tissues. For instance, Cordero et al. performed SFK gain and loss of function experiments in mouse and fruit fly intestines to address SFK’s physiological role in the intestinal epithelium [15]. Drosophila is a useful genetic model to study intestinal homeostasis because, as in mammals, adult fly midgut epithelium is renewed by intestinal stem cells (ISC) [16]. Src42A and Src64B are the two Src-like kinases expressed in Drosophila, and are the likely orthologues of Src and Fyn, respectively. However, only Src42A loss of function inhibits ISC proliferation in conditions of homeostasis and stress response to bacterial infection, suggesting a specific Src function in the intestine. Nevertheless, genetic overactivation of any of these SFKs is sufficient to drive ISC hyperproliferation, indicating potential SFK redundant functions above a certain threshold [15][17]. An Src key role was confirmed in a mouse model where Src ablation in the intestine prevented ISC proliferation and crypt regeneration after induction of DNA damage by gamma irradiation [15]. However, Src ablation alone was not sufficient to affect intestinal homeostasis because of its overlapping functions with Fyn and Yes. In agreement, combined inactivation of Src, Fyn and Yes in the intestine leads to intestinal epithelial cell (IEC) apoptosis and reduction of the number of Paneth cells in the small intestine [15], Conversely, ablation of their negative regulator Csk increases IEC proliferative activity and turnover [18] (Figure 1).
Mechanistically, Src drives ISC proliferation through upregulation of EGFR, activation of Ras/MAPK and signal transducer and activator of transcription 3 (Stat3) signalling [15]. This Src function was revealed after intestinal injury induced by irradiation. The mechanism underlying Src-mediated ISC proliferation is not fully clear, but may implicate important intestinal regulators, such as Wnt/beta-catenin signalling [15], that controls proliferation and differentiation of crypt-localised ISCs, and Notch that controls the enterocyte lineage [19] (Figure 1B). Src also uses the transcription factor Yes Associated Protein (YAP), an essential sensor of the cell microenvironment structural and mechanical features [20], to mediate epithelial regeneration during intestinal inflammation. This new Src signalling activity was uncovered from a genetic mouse model with persistent intestinal inflammation upon IEC overexpression of gp130, the coreceptor for interleukins of the IL-6 family [21]. These animals display aberrant IEC proliferation and differentiation and are resistant to mucosal erosion. This gp130 activity is mediated by interaction with Src and Yes to phosphorylate the transcription factor YAP on specific tyrosine residues and to induce its stabilisation and nuclear translocation [22]. Surprisingly, this inflammatory mechanism is independent from the effector Stat3 [21]. A similar Src/YAP signalling has been described during intestinal regeneration mediated by dietary and metabolic factors [23] strengthening the conserved role of Src/YAP signalling in intestinal repair. Src may also induce ISC proliferation through a cell nonautonomous mechanism. Indeed, in Drosophila gut, upon bacterial infection, Src activation in enterocytes induces IL-6 expression that leads to ISC proliferation [24]. Similarly, in the mouse, in physiological conditions, intestinal tissue is renewed by Leucine-rich repeat-containing G-protein coupled receptor 5-positive (Lgr5+) ISCs localised at the bottom of the crypt [25]. However, upon severe damage, intestinal epithelium can be regenerated by a distinct mechanism [26] that involves IEC dedifferentiation via transient foetal-like features [27]. Importantly, this regenerative mechanism is mediated by extracellular matrix remodelling that enables biomechanic Src/YAP signalling for efficient tissue repair [27] (Figure 1B). A similar mechanism was reported for Class A basic helix-loop-helix protein 15-positive (Bhlha15+) intestinal secretory precursors that transiently convert into enterocyte progenitors after doxorubicin-induced epithelial injury [19]. In the mouse, elevated SFK activity induced by Csk ablation in IECs activates an additional Rac signalling mechanism to promote IEC proliferation [18]. Interestingly, these animals display enhanced susceptibility to colitis induced by the chemical irritant dextran sodium sulphate (DSS), due to low epithelial barrier function caused by tight junctions reduction [28].
Finally, SFKs have also specific functions in immune cells. Indeed, in Fyn-deficient mice exposed to DSS to induce intestinal injury, the number of CD4+FOXP3+ cells was reduced as well as the capacity of lymphocytes to differentiate into regulatory T cells [29]. This indicates that Fyn has a protective role against intestinal injury. Using a similar approach it was demonstrated that Lyn also is protective against intestinal injury, microbiota-dependent intestinal inflammation and susceptibility to enteric pathogens [30]. Another mouse strain with an activated Lyn mutation (LynY508F) revealed a Lyn key role in the control of the innate immune response and validated its protective role against colitis [31].
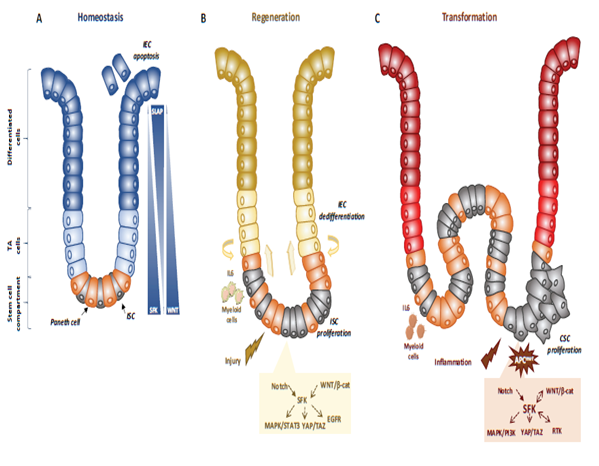
Figure 1. SFKs in intestinal homeostasis, regeneration and transformation. (A) SFKs regulate ISC proliferation, Paneth cell differentiation and IEC survival during intestinal homeostasis. SFKs, Wnt and Slap activity in the intestinal epithelium is indicated. (B) SFKs mediate intestinal regeneration by activating ISC proliferation. (C) SFKs mediate tumour formation induced by activating CSC survival. Src signalling involved in intestinal regeneration and transformation is indicated.
3. SFKs in Intestinal Cell Transformation
- SFKs in Intestinal Cell Transformation
Intestinal transformation is mainly caused by deregulated ISC proliferation [32], that can be mediated by SFKs (Figure 1C) [21]. Their transforming activity was revealed using genetically-modified animal models [21]. In CRC, the most frequent tumour-initiating event is abnormally elevated Wnt/beta-catenin signalling. Mechanistically, Apc inactivation leads to protein stabilization and activation of beta-catenin transcriptional activity [33]. Additionally, Apc inactivation leads to upregulation of SFK activity in the hyperproliferative “crypt progenitor cell-like” domain of the intestinal epithelium [21][32]. Src gene inactivation in IECs revealed its essential role in intestinal tumorigenesis-induced by Apc loss in mouse and fly models [21]. Specifically, conditional Src inactivation in IECs impairs adenoma tumour initiation and progression. However, this Src activity is not mediated by MAPK or STAT3 signalling, contrary to what is observed during intestinal regeneration [21]. In agreement, Apc inactivation also induces local inflammation due to a reduction in intestinal epithelial barrier function, which also contributes to tumour development [34]. Mechanistically, colon microbiota invasion activates IL-23-producing myeloid cells and expand tumour-resident IL-17-producing T lymphocytes, resulting in proliferation of transformed IECs through IL-11 receptor and gp130 upregulation, and eventually gp130/Src/YAP signalling [35][36]. Importantly, a positive autoregulatory loop controls gp130 and YAP expression, enabling tumour development. In agreement, in transgenic mice that express an active gp130 mutant in IECs, intestinal tumour development is accelerated upon Apc inactivation. Importantly, this gp130 pro-tumoral function depends on SFK activity [36]. The intestinal tumour-promoting role of other SFKs is largely unknown. Nevertheless, a recent study uncovered an important HCK function in colon tumorigenesis [37]. They observed that mutant mice carrying an oncogenic mutation in the Hck gene (HCKY520F) are prone to colon cancer development when treated with DSS and the carcinogen azoxymethane (AOM). This tumour-promoting effect was associated with HCK capacity to induce tumour-promoting M2-like macrophages and the accumulation of IL-6/lL-11 family cytokines [37].
4. Therapeutic Strategies to Target SFKs Signalling in CRC
- Therapeutic Strategies to Target SFKs Signalling in CRC
4.1. Therapeutic Utility
Accumulated evidences obtained in experimental CRC models suggest that SFKs could be attractive targets in advanced CRC. SFK inhibition may be of clinical value in advanced CRC due to their role in CRC cell dissemination and CSC activity, which are the main causes of tumour relapse and metastatic progression. In agreement, specific Src inhibitors (Srci) reduce liver metastasis development in nude mouse models, an effect associated with decreased tumour angiogenesis, cell proliferation and survival [38][39]. However, it was not assessed whether SFK inhibition reduces CSC activity. In addition, Src oncogenic role in RTK signalling may explain why Srci sensitises CRC to RTK inhibitors in experimental CRC models [40]. Similarly, the effect of Src activity on MAPK/PI3K signalling is consistent with findings showing the potential clinical utility of combining Srci with KRAS effector inhibitors (MAPK kinase, and PI3K inhibitors) in KRAS mutant CRC [41][42]. This observation is clinically relevant because these tumours are refractory to the upstream EGFR antibody currently used in the clinic, cetuximab [43]. Src activity has been identified as a mechanism of tumour resistance to oxaliplatin in metastatic CRC, suggesting that its pharmacological inhibition could enhance the efficacy of oxaliplatin-based chemotherapy in patients with CRC [44][45]. Finally, some evidences suggest that Srci might sensitise CRC to anti PD-L1 immune checkpoint inhibitors [46][47].
4.2. Therapeutic Strategies
Several Src-like ATP competitive inhibitors have been developed for oncology, including dasatinib, bosutinib and saracatinib [48]. Although dasatinib and bosutinib were originally developed to target Src/Abl activities, they are multikinase inhibitors [49][50][51]. Moreover, tirbanibulin, a Src-like peptide binding site inhibitor, inhibits also tubulin polymerization [52]. Although most of these Srci display significant anti-tumour activity in experiment tumour models, they gave disappointing results in patients with CRC, both as monotherapy and in combination with the current therapies [53][54][55]. For instance, the combination of dasatinib with the chemotherapy regimen FOLFOX (folinic acid, 5-fluorouracil, and oxaliplatin) with or without cetuximab did not show any meaningful clinical activity in refractory CRC [55].
The complex mechanisms of Src regulation and hyperactivation in CRC discussed above may explain the lack of anticancer effect of these drugs, particularly the lack of patient stratification, drug efficacy and selectivity, resulting in significant toxicity. These complex mechanisms also suggest new therapeutic strategies to better target Src signalling in CRC (Figure 2).
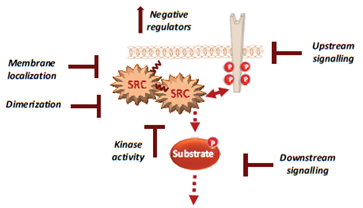
Figure 2. Therapeutic strategies to efficiently target Src signalling in CRC, including drug inhibition of Src UD signalling, kinase dimerisation, membrane localisation, drugs that reactivate Src inhibitors and drugs that inhibit activity of downstream signals.
For instance, several noncatalytic strategies could be developed to improve Src inhibition in CRC, including allosteric inhibitors of the myristoyl switch regulatory mechanism, as recently demonstrated with asciminib in BCR-ABL expressing chronic myeloid leukaemia [56]. Small molecules that interfere with Src UD signalling, kinase dimerisation or membrane localisation by disrupting UNC119-Src interaction may lead to Srci with higher specificity and lower toxicity [57]. Additionally, drugs that reactivate SRC inhibitors, such as PAG or SLAP, could limit CRC invasion or metastatic reactivation. Finally, patient selection based on Src activity level in CRC would clearly improve the overall therapeutic response. While CMS2 tumours should preferentially respond to Srci, studies in advanced CRC suggest that left-sided tumours with elevated RTK signalling are also good targets [58][59]. Moreover, there is no validated biomarker for Src-dependent tumours. Nevertheless, several candidates could be proposed, such as high Src tumour activity or SRC copy number, high phosphorylation of Src effectors, such as FAK, YAP/TAZ, RTKs, or even high tyrosine phosphorylation level. However, high SLAP expression could limit Src oncogenic signalling, and therefore, tumours could be less responsive to Srci, despite their high aberrant Src activity [60]. Therefore, SLAP expression could be an additional predictor of the tumour response to Srci.
5. Conclusions and Perspectives
- Conclusions and Perspectives
Since the first observation of abnormal Src activity in CRC samples [61], much has been learned about SFK physiological and oncogenic functions in the intestine. Although, their oncogenic roles have been underestimated because of the absence of frequent somatic mutations in CRC, there is now strong evidence of their detrimental role in CRC cell invasion. This suggests that Srci could be useful for the management of patients with metastatic CRC. However, Srci clinical utility in CRC has not been demonstrated yet, because of lack of patient stratification, drug efficacy and selectivity. Clearly much more needs to be learned about how SFKs function during CRC development to reach this objective. Recent molecular studies have highlighted the much higher complexity of SFK regulation, which needs to be investigated in order to efficiently target these activities. Future studies on SFK physiological roles in the intestine may bring important insights into SFK influence on CRC development. Moreover, appropriate CRC models are crucially needed, including genetically modified mice, to recapitulate some of the activating mechanisms reported in human CRC, in order to assess the complexity of Src signalling. It would be important to analyse the respective oncogenic roles of these SFKs in the epithelial and microenvironment compartments of these tumours. Moreover, their contribution to metastatic reactivation and immune evasion also are important questions that could be addressed with these models. Finally, phosphoproteomic studies are needed to decipher the molecular complexity of Src signalling in CRC. For instance, the large group of mRNA regulators identified in such studies [62] points to an unsuspected feature of Src tumour activity. Overall, future studies should allow understanding of how SFKs regulate epithelial homeostasis and tumorigenesis, and improving Src-based therapies in CRC.
References
- T.J. Yeatman; A renaissance for SRC. Nature Reviews Cancer 2004, 4, 470-480, 10.1038/nrc1366.
- Lori C. Kim; Lanxi Song; Eric B. Haura; Src kinases as therapeutic targets for cancer. Nature Reviews Clinical Oncology 2009, 6, 587-595, 10.1038/nrclinonc.2009.129.
- Yeatman, T.J.; A renaissance for SRC. Nat. Rev. Cancer 2004, 4, 470–480, doi:10.1038/nrc1366.
- Titus J Boggon; Michael J. Eck; Structure and regulation of Src family kinases. Oncogene 2004, 23, 7918-7927, 10.1038/sj.onc.1208081.
- Masato Okada; Regulation of the Src Family Kinases by Csk. International Journal of Biological Sciences 2012, 8, 1385-1397, 10.7150/ijbs.5141.
- G S Martin; The hunting of the Src. Nature Reviews Molecular Cell Biology 2001, 2, 467-475, 10.1038/35073094.
- Miguel Arbesú; Mariano Maffei; Tiago Cordeiro; João Miguel Correia Teixeira; Yolanda Pérez; Pau Bernadó; Serge Roche; Miquel Pons; The Unique Domain Forms a Fuzzy Intramolecular Complex in Src Family Kinases. Structure 2017, 25, 630-640.e4, 10.1016/j.str.2017.02.011.
- Mariano Maffei; Miguel Arbesú; Anabel-Lise Le Roux; Irene Amata; Serge Roche; Miquel Pons; The SH3 Domain Acts as a Scaffold for the N-Terminal Intrinsically Disordered Regions of c-Src. Structure 2015, 23, 893-902, 10.1016/j.str.2015.03.009.
- Danislav Spassov; Ana Ruiz-Saenz; Amit Piple; Mark M. Moasser; A Dimerization Function in the Intrinsically Disordered N-Terminal Region of Src. Cell Reports 2018, 25, 449-463.e4, 10.1016/j.celrep.2018.09.035.
- Anabel-Lise Le Roux; Maria Antonia Busquets; Francesc Sagués; Miquel Pons; Kinetics characterization of c-Src binding to lipid membranes: Switching from labile to persistent binding. Colloids and Surfaces B: Biointerfaces 2016, 138, 17-25, 10.1016/j.colsurfb.2015.11.013.
- Bhushan Nagar; Oliver Hantschel; M. A. Young; Klaus Scheffzek; Darren Veach; William Bornmann; Bayard Clarkson; Giulio Superti-Furga; John Kuriyan; Structural Basis for the Autoinhibition of c-Abl Tyrosine Kinase. Cell 2003, 112, 859-871, 10.1016/s0092-8674(03)00194-6.
- Sandra W Cowan-Jacob; Gabriele Fendrich; Paul W. Manley; Wolfgang Jahnke; Doriano Fabbro; Janis Liebetanz; Thomas Meyer; The Crystal Structure of a c-Src Complex in an Active Conformation Suggests Possible Steps in c-Src Activation. Structure 2005, 13, 861-871, 10.1016/j.str.2005.03.012.
- Anabel-Lise Le Roux; Irrem-Laareb Mohammad; Borja Mateos; Miguel Arbesú; Margarida Gairí; Farman Ali Khan; João Miguel Correia Teixeira; Miquel Pons; A Myristoyl-Binding Site in the SH3 Domain Modulates c-Src Membrane Anchoring.. iScience 2019, 12, 194-203, 10.1016/j.isci.2019.01.010.
- C A Lowell; Philippe Soriano; Knockouts of Src-family kinases: stiff bones, wimpy T cells, and bad memories.. Genes & Development 1996, 10, 1845-1857, 10.1101/gad.10.15.1845.
- Julia B. Cordero; Rachel A Ridgway; Nicola Valeri; Colin Nixon; Margaret C. Frame; William J Muller; Marcos Vidal; Owen J. Sansom; c-Src drives intestinal regeneration and transformation. The EMBO Journal 2014, 33, 1474-1491, 10.1002/embj.201387454.
- Andreu Casali; Eduard Batlle; Intestinal Stem Cells in Mammals and Drosophila. Cell Stem Cell 2009, 4, 124-127, 10.1016/j.stem.2009.01.009.
- A Kohlmaier; C Fassnacht; Y Jin; H Reuter; J Begum; Devanjali Dutta; B A Edgar; Src kinase function controls progenitor cell pools during regeneration and tumor onset in the Drosophila intestine. Oncogene 2014, 34, 2371-2384, 10.1038/onc.2014.163.
- Shinya Imada; Yoji Murata; Takenori Kotani; Masaki Hatano; Chunxiao Sun; Tasuku Konno; Jung-Ha Park; Yasuaki Kitamura; Yasuyuki Saito; Hideki Ohdan; et al.Takashi Matozaki Role of Src Family Kinases in Regulation of Intestinal Epithelial Homeostasis. Molecular and Cellular Biology 2016, 36, 2811-2823, 10.1128/mcb.00311-16.
- Yoku Hayakawa; Mayo Tsuboi; Samuel Asfaha; Hiroto Kinoshita; Ryota Niikura; Mitsuru Konishi; Masahiro Hata; Yukiko Oya; Woosook Kim; Moritz Middelhoff; et al.Yohko HikibaNaoko HigashijimaSozaburo IharaTetsuo UshikuMasashi FukayamaYagnesh TailorYoshihiro HirataChandan GuhaKelley S. YanKazuhiko KoikeTimothy C. Wang BHLHA15-Positive Secretory Precursor Cells Can Give Rise to Tumors in Intestine and Colon in Mice. Gastroenterology 2019, 156, 1066-1081.e16, 10.1053/j.gastro.2018.11.024.
- Toshiro Moroishi; Carsten Gram Hansen; Kun-Liang Guan; The emerging roles of YAP and TAZ in cancer. Nature Cancer 2015, 15, 73-79, 10.1038/nrc3876.
- Koji Taniguchi; Li-Wha Wu; Sergei I. Grivennikov; Petrus R. De Jong; Ian Lian; Fa-Xing Yu; Kepeng Wang; Samuel B. Ho; Brigid S. Boland; John T Chang; et al.William J. SandbornGary HardimanEyal RazYoshihiko MaeharaAkihiko YoshimuraJessica Zucman-RossiKun-Liang GuanMichael Karin A gp130–Src–YAP module links inflammation to epithelial regeneration. Nature 2015, 519, 57-62, 10.1038/nature14228.
- Joseph Rosenbluh; Deepak Nijhawan; Andrew Cox; Xingnan Li; James Neal; Eric J. Schafer; Travis I. Zack; Xiaoxing Wang; Aviad Tsherniak; Anna C. Schinzel; et al.Diane D. ShaoSteven E. SchumacherBarbara A. WeirFrancisca VazquezGlenn S. CowleyDavid E. RootJill P. MesirovRameen BeroukhimCalvin J. KuoWolfram GoesslingWilliam C. Hahn β-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis.. Cell 2012, 151, 1457-73, 10.1016/j.cell.2012.11.026.
- Giovanni Sorrentino; Alessia Perino; Ece Yildiz; Gaby El Alam; Maroun Bou Sleiman; Antimo Gioiello; Roberto Pellicciari; Kristina Schoonjans; Bile Acids Signal via TGR5 to Activate Intestinal Stem Cells and Epithelial Regeneration. Gastroenterology 2020, S0016-5085(20), 34739-9, 10.1053/j.gastro.2020.05.067.
- Philip Houtz; Alessandro Bonfini; Xi Liu; Jonathan Revah; Aurelien Guillou; Mickaël Poidevin; Korneel Hens; Hsin-Yi Huang; Bart Deplancke; Yu-Chen Tsai; et al.Nicolas Buchon Hippo, TGF-β, and Src-MAPK pathways regulate transcription of the upd3 cytokine in Drosophila enterocytes upon bacterial infection. PLoS Genetics 2017, 13, e1007091, 10.1371/journal.pgen.1007091.
- Nick Barker; Alexander Van Oudenaarden; Hans Clevers; Identifying the Stem Cell of the Intestinal Crypt: Strategies and Pitfalls. Cell Stem Cell 2012, 11, 452-460, 10.1016/j.stem.2012.09.009.
- Cédric Blanpain; Elaine Fuchs; Plasticity of epithelial stem cells in tissue regeneration. Science 2014, 344, 1242281-1242281, 10.1126/science.1242281.
- Shiro Yui; Luca Azzolin; Martti Maimets; Marianne Terndrup Pedersen; Robert P. Fordham; Stine Lind Hansen; Hjalte List Larsen; Jordi Guiu; Mariana Rp Alves; Carsten F. Rundsten; et al.Jens V. JohansenYuan LiChris D. MadsenTetsuya NakamuraMamoru WatanabeOle Haagen NielsenPawel J. SchweigerStefano PiccoloKim B Jensen YAP/TAZ-Dependent Reprogramming of Colonic Epithelium Links ECM Remodeling to Tissue Regeneration. Cell Stem Cell 2018, 22, 35-49.e7, 10.1016/j.stem.2017.11.001.
- Chunxiao Sun; Yoji Murata; Shinya Imada; Tasuku Konno; Takenori Kotani; Yasuyuki Saito; Hideto Yamada; Takashi Matozaki; Role of Csk in intestinal epithelial barrier function and protection against colitis. Biochemical and Biophysical Research Communications 2018, 504, 109-114, 10.1016/j.bbrc.2018.08.140.
- Fernando Lopes; Arthur Wang; David Smyth; Jose-Luis Reyes; Axinia Doering; L. Patrick Schenck; Paul Beck; Christopher Waterhouse; Derek M. McKay; The Src kinase Fyn is protective in acute chemical-induced colitis and promotes recovery from disease. Journal of Leukocyte Biology 2015, 97, 1089-1099, 10.1189/jlb.3a0814-405rr.
- Morgan Roberts; Jennifer L. Bishop; Xueling Fan; Jennifer L. Beer; Winnie W. S. Kum; Danielle L. Krebs; Morris Huang; Navkiran Gill; John J. Priatel; B. Brett Finlay; et al.Kenneth W. Harder Lyn Deficiency Leads to Increased Microbiota-Dependent Intestinal Inflammation and Susceptibility to Enteric Pathogens. The Journal of Immunology 2014, 193, 5249-5263, 10.4049/jimmunol.1302832.
- J L Bishop; Morgan Roberts; J L Beer; M Huang; M K Chehal; X Fan; L A Fouser; H L Ma; J T Bacani; K W Harder; et al. Lyn activity protects mice from DSS colitis and regulates the production of IL-22 from innate lymphoid cells. Mucosal Immunology 2013, 7, 405-416, 10.1038/mi.2013.60.
- Nick Barker; Rachel A. Ridgway; Johan H. Van Es; Marc Van De Wetering; Harry Begthel; Maaike Van Den Born; Esther Danenberg; Alan R. Clarke; Owen J. Sansom; Hans Clevers; et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2008, 457, 608-611, 10.1038/nature07602.
- Roeland Nusse; Hans Clevers; Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985-999, 10.1016/j.cell.2017.05.016.
- Sergei I. Grivennikov; Kepeng Wang; Daniel Mucida; C. Andrew Stewart; Bernd Schnabl; Dominik Jauch; Koji Taniguchi; Guann-Yi Yu; Christoph H. Österreicher; Kenneth E. Hung; et al.Christian DatzYing FengEric R. FearonMohamed OukkaLino TessarolloVincenzo CoppolaFelix YarovinskyHilde CheroutreLars EckmannGiorgio TrinchieriMichael Karin Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 2012, 491, 254-258, 10.1038/nature11465.
- Kepeng Wang; Min Kyoung Kim; Giuseppe Di Caro; Jerry Wong; Shabnam Shalapour; Jun Wan; Wei Zhang; Zhenyu Zhong; Elsa Sanchez-Lopez; Li-Wha Wu; et al.Koji TaniguchiYing FengEric FearonSergei I. GrivennikovMichael Karin Interleukin-17 receptor a signaling in transformed enterocytes promotes early colorectal tumorigenesis.. Immunity 2014, 41, 1052-63, 10.1016/j.immuni.2014.11.009.
- Koji Taniguchi; Toshiro Moroishi; Petrus R. De Jong; Michal Krawczyk; Britta M. Grebbin; Huiyan Luo; Rui-Hua Xu; Nicole Golob-Schwarzl; Caroline Schweiger; Kepeng Wang; et al.Giuseppe Di CaroYing FengEric R. FearonEyal RazLukas KennerHenner F. FarinKun-Liang GuanJohannes HaybaeckChristian DatzKang ZhangMichael Karin YAP–IL-6ST autoregulatory loop activated on APC loss controls colonic tumorigenesis. Proceedings of the National Academy of Sciences 2017, 114, 1643-1648, 10.1073/pnas.1620290114.
- Justin M. Summy; Gary E. Gallick; Src family kinases in tumor progression and metastasis.. Cancer and Metastasis Reviews 2003, 22, 337-358, 10.1023/A:1023772912750.
- Jeong-Seok, Nam; Yoshinori, Ino; Michiie, Sakamoto; Setsuo, Hirohashi; Src family kinase inhibitor PP2 restores the E-cadherin/catenin cell adhesion system in human cancer cells and reduces cancer metastasis. Clinical Cancer Research 2002 , 7, 2430-2436, PMID: 12114449.
- Scott Kopetz; Donald P. Lesslie; Nikolas A. Dallas; Serk In Park; Marjorie Johnson; Nila U. Parikh; Michael P. Kim; James L. Abbruzzese; Lee M. Ellis; Joya Chandra; et al.Gary E. Gallick Synergistic activity of the SRC family kinase inhibitor dasatinib and oxaliplatin in colon carcinoma cells is mediated by oxidative stress.. Cancer Research 2009, 69, 3842-9, 10.1158/0008-5472.CAN-08-2246.
- Emily F. Dunn; Mari Iida; Rebecca A. Myers; David A. Campbell; Kylee A. Hintz; Eric A. Armstrong; Chunrong Li; Deric L. Wheeler; Dasatinib sensitizes KRAS mutant colorectal tumors to cetuximab. Oncogene 2010, 30, 561-574, 10.1038/onc.2010.430.
- Grace R. Anderson; Peter S. Winter; Kevin H. Lin; Daniel P. Nussbaum; Merve Cakir; Elizabeth M. Stein; Ryan S. Soderquist; Lorin Crawford; Jim C. Leeds; Rachel Newcomb; et al.Priya SteppCatherine YipSuzanne E. WardellJennifer P. TingleyMoiez AliMengmeng XuMeagan RyanShannon J. McCallAutumn J. McReeChristopher M. CounterChanning J. DerKris C. Wood A Landscape of Therapeutic Cooperativity in KRAS Mutant Cancers Reveals Principles for Controlling Tumor Evolution. Cell Reports 2017, 20, 999-1015, 10.1016/j.celrep.2017.07.006.
- Sang-Min Park; Chae Young Hwang; Jihye Choi; Chang Young Joung; Kwang-Hyun Cho; Feedback analysis identifies a combination target for overcoming adaptive resistance to targeted cancer therapy. Oncogene 2020, 39, 3803-3820, 10.1038/s41388-020-1255-y.
- Anita Sveen; Scott Kopetz; Ragnhild A. Lothe; Biomarker-guided therapy for colorectal cancer: strength in complexity. Nature Reviews Clinical Oncology 2019, 17, 11-32, 10.1038/s41571-019-0241-1.
- Scott Kopetz; Van Morris; Nila Parikh; Michael J. Overman; Zhi-Qin Jiang; Dipen Maru; Paul Elvin; Gary E. Gallick; Src activity is modulated by oxaliplatin and correlates with outcomes after hepatectomy for metastatic colorectal cancer.. BMC Cancer 2014, 14, 660, 10.1186/1471-2407-14-660.
- Marco Perez; Antonio Lucena-Cacace; Luis Miguel Marín-Gómez; Javier Padillo-Ruiz; Maria Jose Robles-Frias; Carmen Sáez; R. García-Carbonero; Amancio Carnero; Dasatinib, a Src inhibitor, sensitizes liver metastatic colorectal carcinoma to oxaliplatin in tumors with high levels of phospho-Src. Oncotarget 2016, 7, 33111-33124, 10.18632/oncotarget.8880.
- Can Hekim; Mette Ilander; Jun Yan; Erin Michaud; Richard Smykla; Markus Vähä-Koskela; Paula Maria Savola; Siri Tähtinen; Leena Saikko; Akseli Hemminki; et al.Panu E. KovanenKimmo PorkkaFrancis Y.F. LeeSatu Mustjoki Dasatinib changes immune cell profiles concomitant with reduced tumor growth in several murine solid tumor models.. Cancer Immunology Research 2017, 5, 157-169, 10.1158/2326-6066.CIR-16-0061-T.
- Megan M. Tu; Francis Y. F. Lee; Robert T. Jones; Abigail K. Kimball; Elizabeth Saravia; Robert F. Graziano; Brianne Coleman; Krista Menard; Jun Yan; Erin Michaud; et al.Han ChangHany A. Abdel-HafizAndrii I. RozhokJason E. DuexNeeraj AgarwalAna Chauca-DiazLinda K. JohnsonTerry L. NgJohn CambierEric T. ClambeyJames C CostelloAlan J KormanDan Theodorescu Targeting DDR2 enhances tumor response to anti–PD-1 immunotherapy. Science Advances 2019, 5, eaav2437, 10.1126/sciadv.aav2437.
- Stefano Martellucci; Letizia Clementi; Samantha Sabetta; Vincenzo Mattei; Lorenzo Botta; Adriano Angelucci; Src Family Kinases as Therapeutic Targets in Advanced Solid Tumors: What We Have Learned So Far. Cancers 2020, 12, 1448, 10.3390/cancers12061448.
- Marcus Bantscheff; Dirk Eberhard; Yann Abraham; Sonja Bastuck; Markus Boesche; Scott Hobson; Toby Mathieson; Jessica Perrin; Manfred Raida; Christina Rau; et al.Valérie ReaderGavain SweetmanAndreas BauerTewis BouwmeesterCarsten HopfUlrich KruseGitte NeubauerNigel RamsdenJens RickBernhard KusterGerard Drewes Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nature Biotechnology 2007, 25, 1035-1044, 10.1038/nbt1328.
- Uwe Rix; Oliver Hantschel; Gerhard Dürnberger; Lily L. Remsing Rix; Melanie Planyavsky; Nora V. Fernbach; Ines Kaupe; Keiryn L. Bennett; Peter Valent; Jacques Colinge; et al.Thomas KöcherGiulio Superti-FurgaThomas Köcher Chemical proteomic profiles of the BCR-ABL inhibitors imatinib, nilotinib, and dasatinib reveal novel kinase and nonkinase targets. Blood 2007, 110, 4055-4063, 10.1182/blood-2007-07-102061.
- L L Remsing Rix; U Rix; Jacques Colinge; Oliver Hantschel; K L Bennett; T Stranzl; A Müller; C Baumgartner; Peter Valent; M Augustin; et al.J H TillGiulio Superti-Furga Global target profile of the kinase inhibitor bosutinib in primary chronic myeloid leukemia cells. Leukemia 2008, 23, 477-485, 10.1038/leu.2008.334.
- Asal Fallah-Tafti; Alireza Foroumadi; Rakesh Tiwari; Amir Nasrolahi Shirazi; David G. Hangauer; Yahao Bu; Tahmineh Akbarzadeh; Keykavous Parang; A. Shafiee; Thiazolyl N-benzyl-substituted acetamide derivatives: Synthesis, Src kinase inhibitory and anticancer activities. European Journal of Medicinal Chemistry 2011, 46, 4853-4858, 10.1016/j.ejmech.2011.07.050.
- S. M. Reddy; Scott Kopetz; Jeffrey S. Morris; Nila Parikh; Wei Qiao; Michael J. Overman; D. Fogelman; Imad Shureiqi; C. Jacobs; Z. Malik; et al.Carlos A JimenezR. A. WolffJames L AbbruzzeseGary E. GallickCathy Eng Phase II study of saracatinib (AZD0530) in patients with previously treated metastatic colorectal cancer. Investigational New Drugs 2015, 33, 977-984, 10.1007/s10637-015-0257-z.
- Steven J. Isakoff; D Wang; M Campone; Antonio Calles; E Leip; K Turnbull; N Bardy-Bouxin; L Duvillié; E Calvo; Bosutinib plus capecitabine for selected advanced solid tumours: results of a phase 1 dose-escalation study. British Journal of Cancer 2014, 111, 2058-2066, 10.1038/bjc.2014.508.
- C.M. Parseghian; Nila U. Parikh; Ji Yuan Wu; Zhi-Qin Jiang; Laura Henderson; Feng Tian; Brice Pastor; Marc Ychou; Kanwal Raghav; Arvind Dasari; et al.David R. FogelmanAnastasia D. KatsiampouraDavid G. MenterRobert A. WolffCathy EngMichael J. OvermanAlain R. ThierryGary E. GallickScott Kopetz Dual Inhibition of EGFR and c-Src by Cetuximab and Dasatinib Combined with FOLFOX Chemotherapy in Patients with Metastatic Colorectal Cancer.. Clinical Cancer Research 2017, 23, 4146-4154, 10.1158/1078-0432.CCR-16-3138.
- Christopher A. Eide; Matthew S. Zabriskie; Samantha L. Savage Stevens; Orlando Antelope; Nadeem A. Vellore; Hein Than; Anna Reister Schultz; Phillip Clair; Amber D. Bowler; Anthony Pomicter; et al.Dongqing YanAnna V. SeninaWang QiangTodd W. KelleyPhilippe SzankasiMichael C. HeinrichJeffrey W. TynerDelphine ReaJean-Michel CayuelaDong-Wook KimCristina E. TognonThomas O'hareBrian J. DrukerMichael W. Deininger Combining the Allosteric Inhibitor Asciminib with Ponatinib Suppresses Emergence of and Restores Efficacy against Highly Resistant BCR-ABL1 Mutants. Cancer Cell 2019, 36, 431-443.e5, 10.1016/j.ccell.2019.08.004.
- Guillaume Garivet; Walter Hofer; Antonios Konitsiotis; Christian Klein; Nadine Kaiser; Tom Mejuch; Eyad Fansa; Rania AlSaabi; Alfred Wittinghofer; Philippe I.H. Bastiaens; et al.Herbert Waldmann Small-Molecule Inhibition of the UNC-Src Interaction Impairs Dynamic Src Localization in Cells. Cell Chemical Biology 2019, 26, 842-851.e7, 10.1016/j.chembiol.2019.02.019.
- Rona Yaeger; Walid K. Chatila; Marla D. Lipsyc; Jaclyn Hechtman; Andrea Cercek; Francisco Sanchez-Vega; Gowtham Jayakumaran; Sumit Middha; Ahmet Zehir; Mark T.A. Donoghue; et al.Daoqi YouAgnes VialeNancy KemenyNeil H. SegalZsofia K. StadlerAnna M. VargheseRitika KundraJianjiong GaoAijazuddin SyedDavid M. HymanEfsevia VakianiNeal RosenBarry S. TaylorMarc LadanyiMichael F. BergerDavid B. SolitJinru ShiaLeonard SaltzNikolaus Schultz Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell 2018, 33, 125-136.e3, 10.1016/j.ccell.2017.12.004.
- Cédric Leroy; Camille Fialin; Audrey Sirvent; Valerie Simon; Serge Urbach; Joël Poncet; Bruno Robert; Patrick Jouin; Serge Roche; Quantitative Phosphoproteomics Reveals a Cluster of Tyrosine Kinases That Mediates Src Invasive Activity in Advanced Colon Carcinoma Cells. Cancer Research 2009, 69, 2279-2286, 10.1158/0008-5472.can-08-2354.
- Cécile Naudin; Audrey Sirvent; Cédric Leroy; Romain M. Larive; Valérie Simon; J Pannequin; Jean-Francois Bourgaux; Josiane Pierre; Bruno Robert; Frédéric Hollande; et al.Serge Roche SLAP displays tumour suppressor functions in colorectal cancer via destabilization of the SRC substrate EPHA2. Nature Communications 2014, 5, 3159, 10.1038/ncomms4159.
- C A Cartwright; M P Kamps; A I Meisler; J M Pipas; W Eckhart; pp60c-src activation in human colon carcinoma.. Journal of Clinical Investigation 1989, 83, 2025-2033, 10.1172/jci114113.
- Audrey Sirvent; Serge Urbach; Serge Roche; Contribution of phosphoproteomics in understanding SRC signaling in normal and tumor cells. PROTEOMICS 2015, 15, 232-244, 10.1002/pmic.201400162.