Neurogenic inflammation in migraine is mainly characterized by the release of neuropeptides such as CGRP and substance P from the trigeminal nerve, leading to arterial vasodilation, plasma protein extravasation, and mast cell degranulation. The involvement of these neuropeptides in migraine is evident, and pro-inflammatory cytokines or chemokines may be involved in this series of reactions.
- blood-brain barrier
- migraine
- neuroinflammation
- IL-1β
- chemokine
- anakinra
1. Introduction
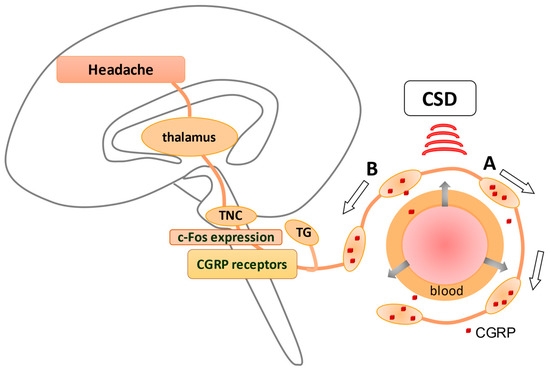
Cortical spreading depression (CSD), relevant to the development of migraine aura, has been shown to activate the trigemino–vascular system (TVS). Anti-temporal conduction of trigeminal ganglion neurons leads to the release of neuropeptides, including calcitonin gene-related peptide (CGRP), from their nerve terminals, leading to vasodilation and plasma extravasation. The dura is densely innervated by sensory nerve fibers that contain these neuropeptides originating from the trigeminal ganglion (TG), causing neurogenic inflammation in the dura (A). In contrast, direct conduction of TG neurons generates a pain sensation via the activation of c-fos in trigeminal nucleus caudalis (TNC) (B), which is eventually perceived as a headache. CGRP receptors are observed in TGs and TNCs but not in peripheral trigeminal nerve terminals, suggesting that CGRP receptor antagonists may suppress neurons in TGs and TNCs. An explanation of how CSD stimulates TVS is given in Figure 1 .
Neurogenic inflammation in migraine is mainly characterized by the release of neuropeptides such as CGRP and substance P from the trigeminal nerve, leading to arterial vasodilation, plasma protein extravasation, and mast cell degranulation. The involvement of these neuropeptides in migraine is evident [1][2][7,8], and pro-inflammatory cytokines or chemokines may be involved in this series of reactions [3][4][9,10].
Neuroinflammation is essentially defined as an inflammatory response in the brain and spinal cord, which is modulated by the production of cytokines, chemokines, reactive oxygen species (ROS), and secondary messengers such as nitric oxide (NO) and prostaglandins [5][6][11,12]. They are mainly produced by activated microglia and astrocytes [5][6][11,12], and neurovascular units comprising neurons, pericytes, and endothelial cells [7][8][13,14]. Neuroinflammation is closely related to central nervous system (CNS) diseases, such as multiple sclerosis and epilepsy, and the regulation of cytokines has revealed new therapeutic aspects.
This review explores the current literature on neuroinflammation. It targets neuroinflammation from the perspective of cytokines in the pathogenesis of migraine and aims to identify new directions for research on therapeutic interventions for migraine targeting neuroinflammation.
2. Clinical Evidence of Neuroinflammation
During migraine attacks (ictal period), interleukin-1β (IL-1β), IL-6, IL-8, and tumor necrosis factor-α (TNF-α) are increased [9][10][11][12][13][14][15,16,17,18,19,20]. Pediatric patients without aura showed higher levels of IL-1β than those with aura [11][17]. However, some reports have shown no significant difference in TNF-α and IL-1β levels between attacks and attack-free periods [15][16][21,22]. Decreased levels of the anti-inflammatory cytokines IL-4 and IL-5 were reported during the attacks [12][17][18,23]. Increased levels of IL-10 (an anti-inflammatory cytokine) during attacks, compared with the interictal period [10][18][19][16,24,25], and a decrease in IL-10 by sumatriptan (one of the triptans) were found [19][25].
During headache-free periods (interictal period), increased pro-inflammatory levels of IL-1β, IL-6, TNF-α, and chemokines such as IL-8, macrophage inflammatory protein-1α (MIP-1α), and C–C chemokine ligand 5 (CCL5) were observed in patients [18][20][21][22][23][24,26,27,28,29]. Conversely, IL-10 levels were found to be similar [9][15] or decreased [19][20][21][25,26,27] when compared with healthy individuals.
While there is some directionality, such as an increase in pro-inflammatory cytokines during both migraine attacks and interictal periods, no definite consensus has been reached. The duration of headache attacks varies widely, which may explain the inconsistent results in terms of sampling timing. Serial analysis of cytokines from jugular venous blood by Sarchielli et al. yielded consistent and robust data which, quite intriguingly, were completely different from data taken from the periphery [24][25][32,33]. Significant increases in TNF-α, IL-6, nuclear factor (NF)-κB, and soluble intercellular adhesion molecule-1 (sICAM-1) were observed in parallel within 2 h of attack onset compared with the time of catheter insertion [25][33]. CGRP increased significantly after 1 h and IL-8 reached its highest level at 4 h, while the other two chemokines, RANTES (CCL5) and monocyte chemoattractant protein-1 (MCP-1), did not significantly change at any time point [24][32]. While a slight increase in IL-1β was observed from 1 to 4 h, the levels of IL-1β decreased, reaching values at the end of the attack [12][18]. The levels of cytokines, selectins, vascular adhesion molecules, and adhesion molecules in the peripheral blood of patients with migraine were found to be unaltered at each time point of the study [24][25][32,33]. The results of these investigations are consistent and significant over time, illustrating the difficulty of examining cytokines in patients with migraine. Carotid artery blood may provide clearer information than peripheral blood regarding biochemical and neurotransmitter changes in the cerebral circulation, and may be more characteristic of migraine attacks.
Despite the normal serum TNF-α levels in patients with chronic migraine (CM), elevated TNF-α levels in the CSF have been identified [26][34]. Although IL-1 receptor antagonist (IL-1RA), MCP-1, and transforming growth factor-β1 were significantly increased in the CSF of patients with episodic tension-type headache and in migraine patients with and without aura compared to those without pain, the increases were not sufficiently different between these headache types [27][35]. A comparison of CSF and plasma or serum cytokine levels would provide some insight into the origin of cytokines, although the assessment of CSF cytokines in patients with migraine is limited. Due to ethical issues, it may be difficult to examine CSF in patients with migraine for the foreseeable future, hence the value of studying available reports.
3. Presence of Blood-Brain Barrier (BBB) Permutability in Migraine
The presence or absence of BBB impairment associated with neuroinflammation is important when considering therapeutic strategies. Studying a drug’s permeability from the periphery to the brain, and its potential to infiltrate cells such as monocytes that introduce cytokines into the brain, will lead to new therapeutic strategies in the future.
Experimental studies of rat models resembling primary headache have shown that CSD modulates the permeability of the BBB by activating matrix metalloproteinase-9 (MMP-9) from 3 h after CSD induction, peaking at 24 h [28][74]. A recent study in awake rats also found that cortical BBB leakage began 0.5 h after CSD induction and resolved within 6 h, without altering the tight junction proteins occludin or claudin-5 [29][75]. CSD-induced BBB opening to water and large molecules is mediated by increased endothelial transcytosis. These phenomena are dependent on caveolin-1 and rho-kinase 2; in contrast, endothelial tight junctions, pericytes, and basement membranes are maintained after CSD [30][76]. This change in BBB permeability is not destructive and may be a transient alteration associated with an inflammatory response. It may also directly affect the access of agents to centrally located targets during migraine attacks [31][77]. Indeed, increased uptake of sumatriptan into the brain was detected, in association with transient BBB permeability, in a KCl-induced model of episodic headache [29][75]. Previous reports suggest that dural inflammation induced by trigeminal ganglion (TG) stimulation does not affect the integrity of the BBB [32][78].
Previous clinical studies have found no association between primary headache and BBB opening [33][79]. Elevations of MMP-9 [34][80] and ICAM-1 [13][19], which are considered typical markers suggestive of BBB disorders, are observed in patients with migraine. An MMP-9 haplotype was reported to affect circulating MMP-9 levels in women with migraine [35][81].
One study reported a significant elevation of endothelial cell-specific molecule-1 (ESM-1) and claudin-5 in the migraine attack group [14][20]. ESM-1 is expressed in the vascular endothelium and is regulated by a number of cytokines, including IL-1β and TNF-α, and growth factors. Claudin-5 regulates BBB permeability and is important in sustaining the integrity of cerebrovascular endothelial cells [36][37][82,83]. A positive correlation was also found between Visual Analog Scale scores and the levels of ESM-1 and claudin-5 [14][20]. In addition to the association of BBB disorders with the pathogenesis of migraine, the results suggest a link between the clinical severity of migraine and BBB disorders.
4. Participation of Pericytes in Migraine
Pericytes are a neurovascular unit component of the BBB and play an important role in the integrity of the BBB. Pericyte degeneration and/or dysfunction contribute to the loss of BBB integrity, which is an early hallmark of several neurodegenerative and inflammatory conditions [7][38][39][13,87,88].
Capillary pericytes are also shown to play an active role in the regulation of cortical vasculature during and after CSD [40][89]. In fact, prolonged vasoconstriction caused by CSD was revealed to be strongest in primary capillaries, where pericytes have a sustained increase in calcium levels. Somatosensory stimulation after CSD causes no further changes in the diameter of capillaries or calcium in pericytes, suggesting that pericytes play a critical role in long-term oligemia after CSD [40][89]. Current research has demonstrated that brain pericytes respond to inflammatory signals, including IL-1β and TNF-α [38][41][42][43][44][87,90,91,92,93]. Pericytes may act as sensors for the inflammatory response in the CNS [43][45][92,94]. Based on BBB integrity and systemic peripheral inflammation, pericytes are speculated to play a pivotal role in the pathogenesis of epilepsy during neuroinflammation [8][14]. Structural changes in pericytes associated with epileptic seizures are exacerbated by IL-1β rather than TNF-α [46][95]. It is uncertain whether migraine, like epilepsy, is associated with BBB disorder and neuroinflammation, and experimental results targeting pericytes have not been obtained. However, structural changes in pericytes and the endothelial cells of microvessels were also observed in patients with familial hemiplegic migraine (FHM) [47][96]. It is known that both migraine and epilepsy share pathologies and clinical features to a certain extent, through similar underlying pathophysiological mechanisms, and antiepileptics such as valproic acid or topiramate are effective in treating patients with both disorders [48][49][97,98]. Therefore, the prevention or treatment of pericyte constriction may become a therapeutic target in migraine [40][50][89,99].