Synovial sarcoma (SS) is a rare and highly malignant tumor and a type of soft tissue sarcoma (STS), for which survival has not improved significantly in recent years. Synovial sarcomas occur mostly in adolescents and young adults (15–35 years old), usually affecting the deep soft tissues near the large joints of the extremities, with males being at a slightly higher risk. Despite its name, synovial sarcoma is neither related to the synovial tissues that are a part of the joints, i.e., the synovium, nor does it express synovial markers; however, the periarticular synovial sarcomas can spread as a secondary tumor to the joint capsule. SS was initially described as a biphasic neoplasm comprising of both epithelial and uniform spindle cell components. Synovial sarcoma is characterized by the presence of the pathognomonic t (X; 18) (p11.2; q11.2) translocation, involving a fusion of the SS18 (formerly SYT) gene on chromosome 18 to one of the synovial sarcoma X (SSX) genes on chromosome X (usually SSX1 or SSX2), which is seen in more than 90% of SSs and results in the formation of SS18-SSX fusion oncogenes.
Note: The following contents are extract from your paper. The entry will be online only after author check and submit it.
1. Introduction
Soft tissue sarcomas in general and synovial sarcoma (SS) in particular are highly malignant tumors, and still represent an unsolved problem for musculoskeletal oncologists in terms of local recurrence and survival rate. Synovial sarcomas of the extremities are the most frequent (80–90%), occurring mostly in adolescents and young adults (15–35 years old, AYA generation), usually affecting the deep soft tissues near the large joints of the extremities, with males being at a slightly higher risk
[1]. The origin of the synovial sarcoma comes not from the articular synovial tissue. However, the occurrence of synovial sarcoma originating from an intraarticular location is suspected to be rare
[2]. The main localizations are in the legs or arms, especially thigh, popliteal fossa and foot, but SS can appear in any part of the body
[3]. The incidence of synovial sarcoma has been estimated to be 2.75 per 100,000, with an incidence of cases by age of 74% for the group aged 20–64 years old and 16% for those 65 years and older
[4].
SS was initially described as a biphasic neoplasm containing both epithelial and uniform spindle cell components. Synovial sarcoma is characterized by the presence of the pathognomonic t (X;18) (p11.2; q11.2) translocation, involving a fusion of the SS18 (formerly SYT) gene on chromosome 18 to one of the synovial sarcoma X (SSX) genes on chromosome X (usually SSX1 or SSX2), which is seen in more than 90% of SSs and results in the formation of SS18-SSX fusion oncogenes
[5,6][5][6].
These tests may include imaging studies, such as an X-ray, CT scan and/or MRI scan; genetic testing to detect the specific chromosomal translocation between chromosome 18 and chromosome X that is identified in most cases of synovial sarcoma; and a biopsy of the tumor
[7]. About 50% of cases showed normal X-rays because of the small size of the lesion or because of the location of the synovial sarcoma in regions with complex neuro-vascular anatomical structures. The CT scan may commonly identify the tumor as a heterogeneous soft-tissue mass with a density that is similar to or slightly lower to that of the muscles. In the evaluation of synovial sarcoma, an MRI exam is the procedure of choice.
Three histological subtypes of synovial sarcomas are described: biphasic (20–30% of lesions); monophasic (50–60%), especially mesenchymal spindle-cell elements; and the poorly differentiated subtype. The lesion is usually well defined and may be surrounded by a pseudocapsule (mainly in small dimension masses), but it also may appear with poorly defined margins and polilobulated, and with areas of intratumoral hemorrhage, necrosis and cystic zones alternating with calcification areas
[8].
Surgery is the mainstay of treatment for synovial sarcoma. The goal is to remove the malignant tissue with a margin of healthy tissue around it. This can sometimes involve the removal of an entire muscle or muscle group, or even amputation. Although treatment is focused on surgery, adjuvant radiation and/or chemotherapy may be beneficial, particularly in high-risk patients and in order to decrease the chances of recurrence. Significant prognostic factors include: size > 5 cm, deep-seated localization, adequacy of surgical margins and history of recurrence
[9].
2. Diagnosis
2.1. Clinical Presentation
Soft tissue sarcomas may appear anywhere in the body, with the majority of them being located at the level of the lower limb, mostly around the knee or hip joint. The nomenclature of synovial sarcoma might be confusing since their origin is not the synovial tissue but the primitive mesenchymal cells
[10].
In the early stages, small synovial sarcomas may cause insignificant signs or symptoms. As the tumor grows larger, the patient may notice a mass or swelling of the affected region. In some cases, the tumor can limit the range of motion or cause numbness and/or pain if it presses on nearby nerves
[11]. The common clinical appearance is a slow-growing painless mass and may give the false impression of being benign. Based on the tumor location, some functional impairment may appear. Sometimes, the symptoms of a synovial sarcoma can be mistaken with other inflammatory conditions, such as bursitis and synovitis, or it may go unnoticed for a long time. Plain radiographs often show small calcifications within the mass. This finding should alert the physician to the diagnosis
[12].
Synovial sarcoma may first be suspected in the presence of characteristic signs and symptoms. Additional tests may be required in order to determine the correct diagnosis and the severity of the condition.
2.2. Imaging Examinations
X-ray: Simple X-rays are not sensitive enough for the diagnosis of SS. Usually, an X-ray is the first imaging study performed as a routine procedure in the presence of an overgrown tumoral mass in an extremity. Calcifications, which can mimic myositis ossificans, and bone involvement (erosions, osteolysis) may be observed on plain X-rays (
Figure 1).
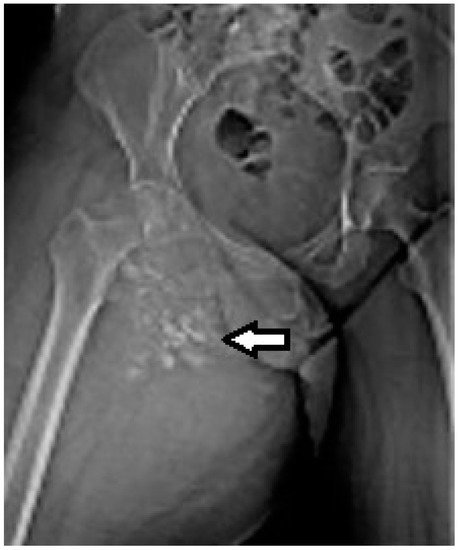
Figure 1. Synovial sarcoma, F, 40 YO—radiological AP view of the right hip and proximal thigh (prior to CT exam) reveals the presence of soft tissue calcifications medial to the lesser trochanter.
Magnetic resonance imaging (MRI) with contrast should be considered for tumors exceeding 2–3 cm and is probably the imaging method of choice for synovial sarcomas. MRI will provide valuable information regarding the extent of the tumor and the relationship with the surrounding anatomical structures; the use of a gadolinium contrast will enhance the amount of information that can be obtained since it is able to differentiate between hemorrhagic areas and solid tumoral tissue.
The T2 sequences are more specific for the diagnosis of soft tissue sarcomas and can reveal an inhomogeneous tumor, mainly with a hyperintense signal, in contrast with muscles, and the specific “triple sign” is present, representing a mixture of solid parts (medium hemorrhage and/or necrosis (high signal intensity) and calcifications or/and fibrotic areas (low signal intensity). The heterogeneity is completed by the presence of cystic parts with an aspect of fluid–fluid levels due to blood sedimentation (
Figure 2 and
Figure 3).
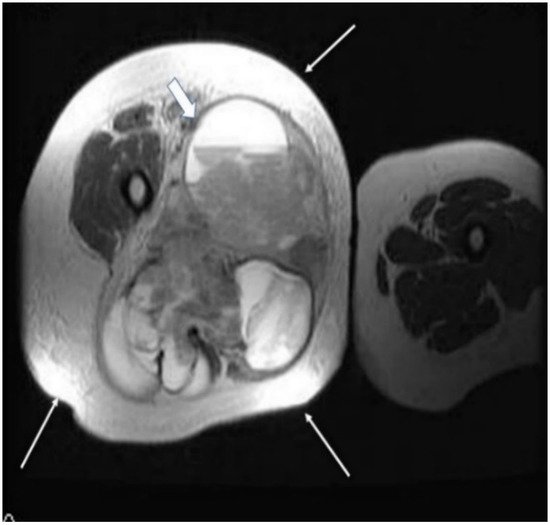
Figure 2. A 40-year old female patient with synovial sarcoma. Image represents an axial MRI of the proximal right thigh—axial T2 FSE (fast spin echo) and FATSAT (fat saturation). There is an increased diameter of the right thigh because of a soft tissue mass (thin arrows), where an inhomogeneous mixture of solid and cystic areas is shown (block arrow).
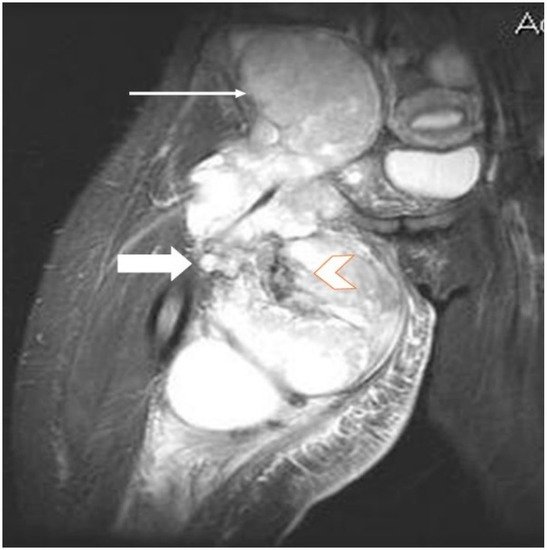
Figure 3. Same patient as in Figure 1 and Figure 2, MR image, coronal T2 STIR (short-time inversion recovery): heterogeneous soft tissue mass showing a mixture of solid areas (line arrow), hemorrhage and/or necrosis (block arrow), calcifications or fibrosis, cystic elements and infiltration of adjacent fat. Furthermore, fluid–fluid levels (block arrow) can be observed in this figure.
Computed tomography (CT) is usually performed either if the MRI is contraindicated (e.g., due to the presence of orthopedic implants), is unavailable or if the radiologists need additional information regarding the local bone involvement or better visualization of the intratumoral calcifications (
Figure 4,
Figure 5 and
Figure 6).
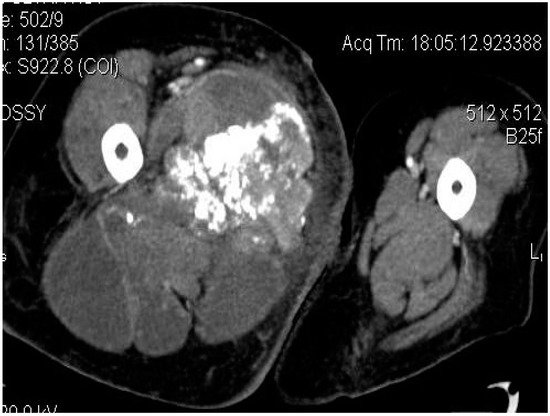
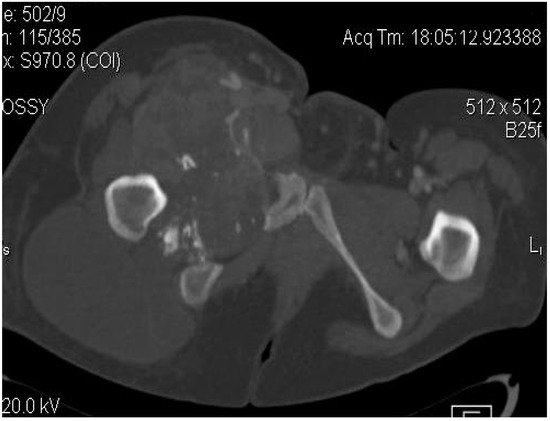
Figure 4. Axial CT-scan: evident enlargement of the right thigh; heterogeneous soft-tissue mass with calcifications, adjacent fat infiltration and skin thickening—synovial sarcoma, M, 26 YO patient.
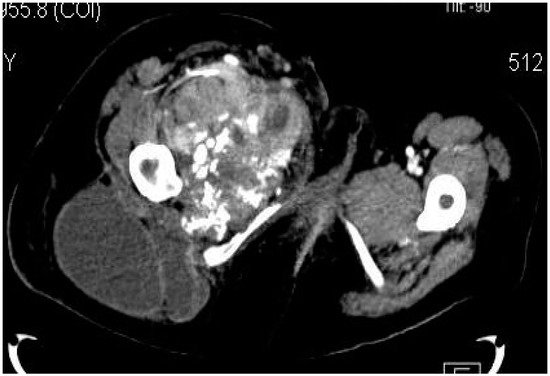
Figure 5. Synovial sarcoma, F, 31 YO patient—CT image, axial section: soft-tissue mass with heterogeneous content, showing necrotic areas alternating with calcifications and cystic areas.
Figure 6. Synovial sarcoma, F, 31 YO patient, CT image, axial section, with contrast—bone window: osteolytic area in the right ischio-pubic rami.
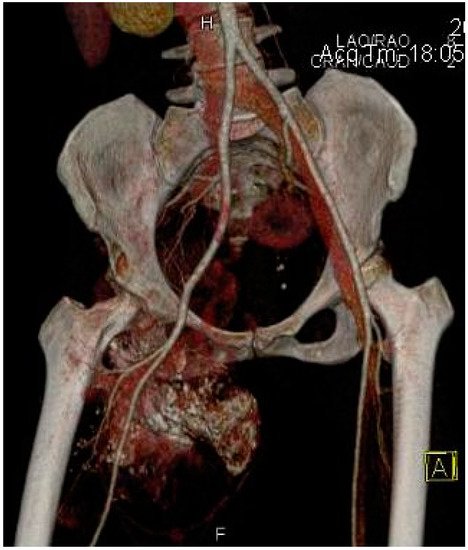
Other imaging examinations may be required. For example, a bone scan may be useful in detecting possible metastatic lesions or an Angio-CT may be requested by the oncologic surgeon, especially for deep synovial sarcomas in order to understand the relationship of the tumor with the neurovascular structures for appropriate preoperative planning (
Figure 7).
Figure 7. Angio-CT with 3D reconstruction indicating the relationship of the synovial sarcoma with the major arterial vessels and the tumoral vascularization.
2.3. Biopsy
A biopsy is mandatory for a proper diagnosis in patients with synovial sarcoma; moreover, it will allow the morphopathologist to differentiate this tumor, especially from other soft tissue sarcomas, and to define the tumor staging.
In order to harvest a representative sample of tumoral tissue, there are several options available, including: a fine needle aspiration, a core needle biopsy (with or without imaging guidance) and an incisional biopsy.
At this point, a core needle biopsy is considered to probably be the best choice, especially if it is performed under ultrasonographic or tomographic guidance for deep, nonpalpable tumors. The major advantage of a core needle biopsy is the amount of tissue that can be retrieved compared to a fine needle aspiration and the small rate of complications compared to an incisional biopsy
[11,13][11][13].
An incisional biopsy should be performed by a musculoskeletal oncologist or after consulting them, where the size of the incision should be as small as possible and it should be in line with the planned incision for the tumoral excision. A good surgical technique is required with careful hemostasis in order to avoid hematoma formation.
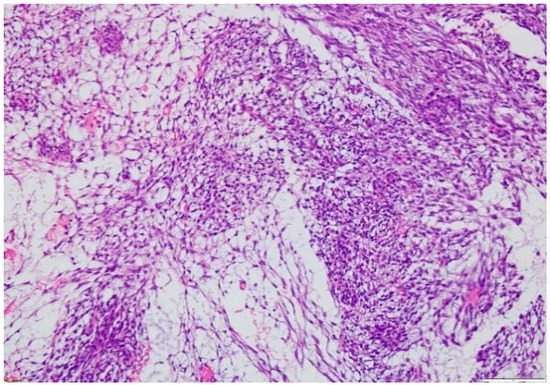
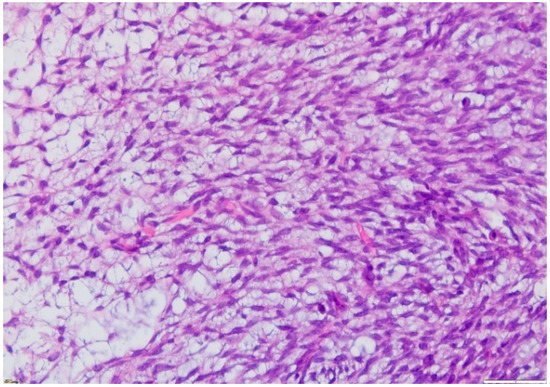
The microscopic exam of the synovial sarcoma will indicate one of the three possible variants: monophasic, biphasic or poorly differentiated. For the first variant, the cellularity is represented by spindle cells, whereas in a biphasic synovial sarcoma, there may also be epithelial cells present (
Figure 8 and
Figure 9). Furthermore, the microscopic exam may reveal the presence of calcifications/ossification areas in up to one-third of cases.
Figure 8. Microscopic findings—classical synovial sarcoma with two main subtypes: biphasic and monophasic appearance, and with two typical cell types: spindle cells and epithelial cells (fusocellular proliferation with myxoid areas, detail), which is rare (HE staining, 20×).
Figure 9. Microscopic finding—monophasic subtype of a synovial sarcoma with mesenchymal spindle cell elements and fusocellular proliferation (HE staining, 40×).
3. Treatment
For the vast majority of the lower limb synovial sarcomas, surgical treatment and adjuvant radiotherapy represent the gold standard of treatment.
3.1. Surgical Treatment
In the past, the main surgical procedure that was used for the removal of a soft tissue sarcoma of the lower limb was amputation, which provided low recurrence rates but sacrificed functional integrity.
The recent evolution of the surgical techniques, together with the adjuvant methods, especially radiotherapy, made the so-called conservative surgery possible for the treatment of synovial sarcomas of the extremities. A wide surgical excision with negative margins is the procedure of choice. Negative margins are extremely important in order to decrease the rate of local recurrence and the overall survival rate. No specific guidelines exist regarding the ideal dimension of the margin that should be resected around the tumoral mass for synovial sarcoma. Usually, for small (<5 cm), superficial tumors, a wide excision with negative margins of approximately 1–2 cm is considered sufficient. In large synovial sarcomas with difficult localizations, achieving a proper negative margin without affecting important neurovascular structures may be difficult. In these situations, very close or microscopically positive margins may occur and the use of adjuvant therapies becomes mandatory in order to avoid local recurrences.
Several methods of tissue reconstruction are available now in order to preserve a satisfactory functional status of the limb following excision.
Use of non-vascularized or vascularized bone grafts, bulk allografts and oncologic endoprosthetic implants are now available for skeletal reconstruction following large osteoarticular resections.
Sacrificing the major vessels may be required for sarcomas that are surrounding these structures. Following this, arterial reconstruction with an autologous saphenous vein or synthetic grafts is mandatory in order to maintain the viability of the limb.
Soft tissue reconstruction may also be required following the excision of synovial sarcoma. Several technical options are available in the surgeon’s armamentarium, such as skin grafts, loco-regional and free flaps for coverage of the residual defect or tendon and muscle transfers for the restoration of motor function.
Coordinated multidisciplinary surgical work is usually needed, especially for large sarcomas with problematic localizations in order to achieve a satisfactory resection, together with a good functional result and a satisfactory cosmesis of the limb.
3.2. Radiotherapy
Adjuvant radiation therapy was shown to facilitate local control in the management of synovial sarcomas. It is usually recommended for high-grade, larger tumors (>5 cm). Radiotherapy may be administered pre- or postoperatively, with a lack of a generally accepted protocol at this time. The timing of radiotherapy is still controversial, with advocates of both pre- or post-surgical radiation. Several complications, such as a fracture, fibrosis, edema and joint stiffness, are reported following radiation therapy for synovial sarcomas
[14].
In order to reduce the volume of radiation and the incidence of complications, intensity-modulated radiation therapy was proposed for adjuvant treatment of soft tissue sarcomas. This technique allows for the use of a higher dose of radiation closer to the core of the tumor, which will reduce the amount of exposure for the surrounding normal tissues.
3.3. Chemotherapy
The use of adjuvant chemotherapy in the treatment of soft tissue sarcoma in general and synovial sarcoma in particular is controversial. Unlike other sarcomas, it seems that there are some subgroups of synovial sarcomas that might benefit from systemic therapy with cytotoxic chemical agents. Usually, chemotherapy is reserved for high-risk, large and deep tumors that have at least an approximately 50% risk of developing metastatic lesions. Based on the existing literature, there is no indication for perioperative chemotherapy in young or adolescent patients with a small SS (<5 cm) when appropriate oncologic margins can be obtained following surgical excision
[15].
Preoperative chemotherapy might also be beneficial by reducing the size of large tumors and causing necrosis and hence allow for easier and complete resection of the tumoral tissue.
However, there are several randomized studies that showed limited or no overall survival benefit with adjuvant chemotherapy
[12,16][12][16].
3.4. Other Therapeutical Options
There is evidence of angiogenesis playing a critical role in the progression of soft tissue sarcomas and, hence, the anti-angiogenic factors might be useful in reducing the tumoral mass. Tyrosine kinase inhibitors, such as endothelial growth factor receptor inhibitors (e.g., sorafenib, pazopanib and sunitinib), were investigated in clinical trials. The most suitable candidates that are considered useful in the treatment of synovial sarcoma might be sorafenib and pazopanib
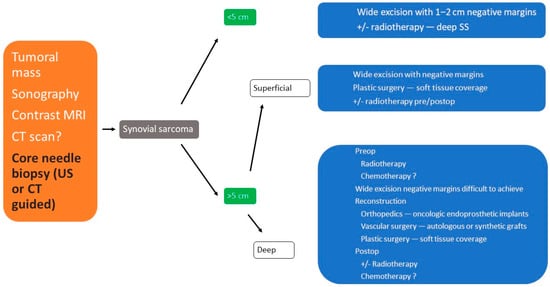
[9,17,18][9][17][18]. (
Figure 10)
Figure 10. Therapeutic algorithm in synovial sarcoma of extremities.
The role of immunotherapy, especially in the treatment of metastatic soft tissue sarcomas, was evaluated. Synovial sarcomas have a high expression of cancer/testis antigen, especially NY-ESO-1, which is highly immunogenic. The safety and activity of immunotherapy with genetically engineered targeted T-lymphocytes to this antigen (autologous T cells expressing NY-ESO-1
c259, which is an affinity-enhanced T-cell receptor (TCR) that recognizes an HLA-A2-restricted NY-ESO-1/LAGE1a-derived peptide) in patients with metastatic synovial sarcoma was evaluated and indicated a clinically significant antitumoral effect
[10,19][10][19].