Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Vivi Li and Version 1 by Deepak Nagrath.
Reactive oxygen species (ROS) are important signaling molecules in cancer. The level of ROS will determine physiological effects. While high levels of ROS can cause damage to tissues and cell death, low levels of ROS can have a proliferative effect. ROS are produced by tumor cells but also cellular components that make up the tumor microenvironment (TME).
- ROS
- mitochondria
- tumorigenesis
- tumor microenvironment
- stroma
- tissue infiltrating lymphocytes
- metabolism
1. Introduction
Reactive oxygen species (ROS) include superoxide, hydrogen peroxide and hydroxyl radicals. While ROS can be damaging to lipids, proteins and DNA, in recent years their role as important intracellular and extracellular signaling molecules has become evident [1]. The mitochondria are the major source of ROS within a cell and play an essential role in regulation of proliferative, apoptotic and metabolic pathways [2,3,4][2][3][4]. It is established that the hallmarks of cancer include metabolic reprogramming as well as a tumor promoting microenvironment [5]. At the interface of both of these important biological events are ROS which are produced by cancer cells as well as cellular components in the microenvironment [6,7,8,9][6][7][8][9]. Understanding how the crosstalk between both extracellular and intracellular ROS not only within the tumor but also with regards to cells that make up the tumor microenvironment (TME) will be critical to our understanding of the process of tumorigenesis.
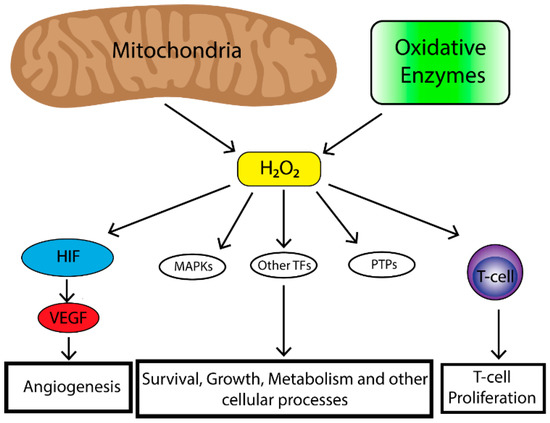
2. Role of Reactive Oxygen Species (ROS) in Tumorigenesis
The majority of endogenous ROS produced in cells result from metabolic reactions occurring within the mitochondria or peroxisome. However, there is a subset of ROS that are also produced by nicotinamide adenine dinucleotide phosphate (NAPDH) oxidases (NOX) which are a family of transmembrane proteins that transport electrons across biological membranes and catalyze the conversion of oxygen into superoxide. Superoxide is then further reduced by superoxide dismutases (SODs) to produce H2O2. ROS can also be produced from cyclooxygenases, lipoxygenases and thymidine phosphorylase [10]. ROS play an important role in tumorigenesis and affect multiple biological processes such as cell proliferation, genomic instability, inflammation, resistance to apoptosis and metabolic reprogramming. Increased levels of ROS are observed in a number of cancer cell lines [11]. In a tumor cell, ROS are primarily generated by the mitochondria. Mitochondria produce superoxide (O2•) from one-electron reduction of oxygen through the mitochondrial electron transport chain (ETC) [12]. Within the mitochondria, ROS are generated at a number of different sites, the most important being complexes I, II and III [13,14][13][14]. Complex I and II generate O2• in the mitochondrial matrix while complex III produces O2• in both the matrix and intermembrane space [15,16][15][16]. O2• generated in the mitochondrial matrix is converted to H2O2 by superoxide dismutase protein 2 (SOD2) [17]. Complex III-generated intermembrane space O2• can travel to the outer mitochondrial membrane and into the cytosol where it is converted into H2O2 by superoxide dismutase protein 1 (SOD1) [18]. Given access to the cytosol, it is thought that complex III-generated ROS are responsible for affecting cellular signaling [19]. As mentioned previously, ROS levels are often elevated in cancer, however, high levels of ROS can have deleterious effects therefore, cells have evolved mechanisms in order to maintain a proper balance of ROS. These mechanisms include peroxide scavenging systems (peroxidases) which control H2O2 levels by reducing H2O2 to H2O [20]. Production of mitochondrial ROS are also regulated by the availability of O2, the rate of electron flux through the ETC, the concentration of given electron carriers and the mitochondrial membrane potential [12,21][12][21]. Finally, the localization of mitochondria within the cell has an important effect on influencing cell signaling pathways as clustering of mitochondria to discrete areas of a cell can preferentially affect adjacent signaling pathways [22,23][22][23]. Together these mechanisms allow for balance of the effects of ROS which can be exploited by tumor cells in order to drive cells preferentially towards a proliferative state. Mitochondrial ROS can stimulate multiple signaling pathways. Perhaps the most well-known is the requirement of mitochondrial ROS for the stabilization of hypoxia-inducible transcription factors (HIFs) under hypoxia [24,25,26,27][24][25][26][27]. HIF stabilization leads to initiation of a broad transcriptional program including regulation of genes important for angiogenesis [28]. In order for angiogenesis to occur proliferation of endothelial cells is required [29]. To that end, HIF upregulates the expression of vascular endothelial growth factor (VEGF). VEGF is a soluble growth factor that binds to VEGF receptors and activates signaling pathways important to endothelial cell proliferation [30]. The mitogenic effects of VEGF are mediated most commonly through the activation of the extracellular-signal-regulated kinase/mitogen-activated protein kinase (ERK/MAPK) pathway which is a potent stimulator of cell proliferation [31]. Mitochondrial ROS are also critical in the activation of T-cells [32]. Reduced levels of complex III-generated mitochondrial ROS in mice leads to the inability for sustained T-cell activation despite stimulation with CD3 or CD28 [32]. Furthermore, a study demonstrated that mitochondria translocate to the immunological synapse in a T-cell line, and mitochondrial H2O2 is required for T-cell receptor (TCR) signal transduction through MAPK signaling [33]. Together, this data suggests that mitochondrial ROS augment TCR signal transduction after antigen stimulation required for T-cell stimulation and proliferation. Together, mitochondrial ROS play an important role in stimulating physiological cell proliferation and can be exploited by a tumor to promote survival and growth. Tumors produce high levels of ROS [11]. Initially it was felt that high levels of ROS contributed to tumorigenesis by oxidative damage to DNA leading to genomic instability [34]. However, studies also demonstrated increased protein expression of cellular antioxidants in cancer cells [35]. Thus, cancer cells have the ability to maintain elevated mitogenic signaling without incurring substantial oxidative damage. Indeed, oncogenes and/or tumor suppressor loss in cancer cells lead to ROS production. For example, a study in which oncogenic H-RasG12V was overexpressed in 3T3 fibroblasts demonstrated increased production of ROS required for mitogenic signaling [36]. Furthermore, mouse embryonic fibroblasts transformed by the loss of p53 tumor suppressor as well as expression of oncogenes Akt, H-RasG12V or KrasG12D demonstrated that mitochondrial ROS are required for anchorage-independent growth in soft agar [6]. Mitochondrial DNA mutations in several genes important for the function of the ETC are present in a number of human cancers [37]. These mutations also lead to increased levels of mitochondrial ROS production [38,39,40][38][39][40]. Loss of mitochondrial transcription factor A (TFAM) in a mouse model of K-ras driven lung cancer demonstrated reduced tumor growth [6]. TFAM is necessary for mitochondrial DNA replication. When TFAM is absent oxidative phosphorylation is impaired and hence levels of mitochondrial ROS are decreased [6]. Furthermore, this study demonstrated that mitochondrial ROS are required for anchorage independent growth in numerous cancer cell types [6] Taken together, the production of ROS by tumor cells plays an important role in driving tumorigenesis however, ROS production by other non-tumor infiltrating cells as well as the overall oxidative state of the local TME has profound effects on tumor biology (Figure 1).Figure 1. The mitochondria are the major contributor to cellular reactive oxygen species (ROS) levels while oxidative enzymes (e.g., NAPDH oxidases, cyclooxygenases, lipooxygenases and thymidine phosphorylase) also contribute to cellular ROS pooles. Mitochondrial ROS have many effects on cellular biology including, Mitogen-activated protein kinase (MAPK) (e.g., extracellular-signal-regulated kinase (ERK), p38 MAPK, Jun N-terminal kinase (JNK)), induction of transcription factors (e.g., nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κβ), hypoxia-inducible transcription factors (HIF), activator protein 1 (AP-1), nuclear respiratory factor (NRF), heat shock factor 1 (HSF-1)) and deregulation of protein phosphatases (e.g., phosphatase and tensin homolog (PTEN)). This leads to enhancement of angiogenesis in the case of HIF, survival, growth, altered metabolism and other cellular processes through MAPKs, transcriptional factors and protein phosphatase and immune cell function and regulation.
3. Cancer-Associated Fibroblasts, ROS and the Tumor Microenvironment (TME)
The TME includes not only tumor cells but tumor lymphatics, tumor vessels, extracellular matrix, non-cancer stromal cells as well as chemical modulators (i.e., chemokines, cytokines, growth factors) and microbial populations. The extracellular matrix (ECM) and stroma include interstitial matrix as well as the basement membrane, and can act as a storage site for many growth factors and chemokines that can stimulate tumorigenesis. Non-cancer stromal cells include endothelial cells, pericytes, immune cells, activated adipocytes, mesenchymal stem cells (MSCs), normal fibroblasts and CAFs. Normal fibroblasts are responsible for ECM turnover and tissue homeostasis. They are fundamental in the processes of wound healing and senescence. Unlike normal fibroblasts, CAFs can be found at the margins of tumors or infiltrating into a tumor. Activated fibroblasts that are found in association with cancer cells are known as CAFs and play key roles in cancer initiation, progression and metastasis [41,42][41][42]. CAFs are further subdivided into fibroblasts and myofibroblasts. Alpha-smooth muscle actin (α-SMA)-positive myofibroblasts are noted to be the major population of CAFs present in tumors [43].
The major role of CAFs is to augment tumorigenesis [44]. Infiltrating CAFs are more proliferative than normal fibroblasts and activate specific signaling pathways important for the promotion of tumor growth [45,46,47][45][46][47]. CAFs residing at the margins of tumors but not within are characterized by their ability to promote cancer progression in vivo [48]. These CAFs are known to secrete factors such as CXCL12 which can go on to activate pro-tumorigenic pathways such as AKT in adjacent epithelial cells [49]. CAFs are present in almost all solid tumors. In certain tumors such as breast, pancreatic and prostate, CAFs can account for up to 80% of the tumor mass as they are responsible for the excessive growth of fibrous or connective tissue (desmoplasia) [50]. A high percentage of CAFs within cancer tissues is associated with poorer prognosis, increased infiltration of tumor-associated macrophages and epithelial to mesenchymal transition (EMT) [50].
Desmoplasia is a marker of tumor progression and generates mechanical forces which can limit the lymphatic and blood supply to a tumor through compression of vessels in turn creating a hypoxic environment [51]. Furthermore, these mechanical forces can cause conversion of fibroblasts to myofibroblasts [51]. As previously mentioned, hypoxia stimulates the production of mitochondrial ROS and cancer cells produce higher levels of ROS than normal tissues which can influence CAF function [9,52][9][52]. CAFs can derive from epithelial, endothelial, hematopoietic stem cells, pericytes or adipocytes as well as resident fibroblasts present in stromal tissue [53,54,55,56,57,58,59][53][54][55][56][57][58][59]. A large proportion of CAFs identified in aggressive adenocarcinomas express smooth-muscle α-actin (α-SMA) and, therefore, are called myofibroblasts [60]. Myofibroblasts’ major function is in wound healing and repair and in tumors these cells can act as drivers for deranged chronic wound healing. Several studies have demonstrated that ROS can be a driver for myofibroblast differentiation. Several studies reported the importance of ROS in the fibroblast to myofibroblast transition (Figure 2A). Transforming growth factor beta 1 (TGF-β1) as well stromal cell-derived factor 1 (SDF-1) and others play a major role in driving the transition from fibroblast to myofibroblast. It is well known that mitochondrial-ROS are required for TGF-β1 activation. Indeed, when fibroblasts were exposed to a pharmacologic inhibitor of mitochondrial-ROS, TGF-β1 expression levels were reduced [61]. Fibroblast to myofibroblast conversion can also be induced with SDF-1 in an ROS-dependent manner [58,60][58][60]. Furthermore, fibroblasts exposed to chronic oxidative stress can also differentiate into myofibroblasts [9,60][9][60]. Fibroblasts isolated from mouse models of oxidative stress in which key antioxidant transcription factors were depleted demonstrated a conversion to myofibroblasts which could be reversed with the long-term treatment with exogenous antioxidants [60,62][60][62]. Additionally, decreased ROS levels due to upregulation of antioxidant enzymes such as glutathione peroxidase 3 and thioredoxin reductase I within fibroblasts from prostate cancer inhibits differentiation into myofibroblasts [63]. Taken together these observations demonstrate that ROS can promote myofibroblast differentiation in human tumors.
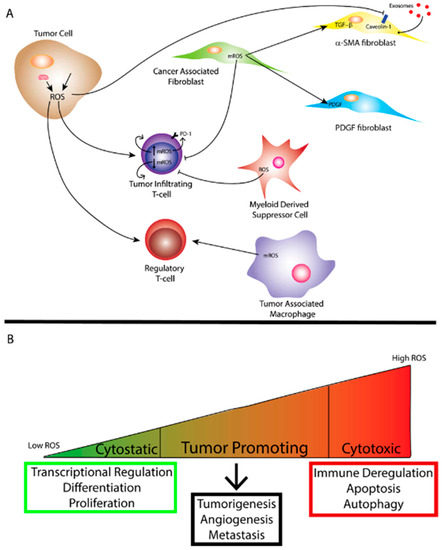
Figure 2. (A) Reactive oxygen species (ROS) generated by the mitochondria and/or exogenous sources within a tumor cell affect tumor immunity to promote a more tumorigenic environment. Mitochondrial ROS (mROS) can stimulate differentiation of cancer-associated fibroblasts (CAFs) and ROS produced by the tumor cell can facilitate uptake of exosomes through caveolin-1 inhibition leading to metabolic reprogramming of certain CAFs. ROS can also affect the function of tumor-infiltrating T-cells depending on the level of mROS. Myeloid-derived suppressor cells (MDSCs) and tumor-associated Macrophages (TAMs) also produce ROS that can affect the function of other immune cells and ROS can affect regulatory T-cell function as well. (B) The amount of ROS corresponds to differing effects on biological function. While cytostatic levels of ROS lead to maintenance of biological processes, cytotoxic levels of ROS lead to cell death as well as immune deregulation. Tumor promotion through ROS occurs when ROS reach super-physiological or cytostatic levels while avoiding levels conducive to cell death. As mentioned previously, oxidative stress can arise from tumor cells.
ROS can also affect proliferation and migration of CAFs. As discussed previously, CAFs exist as a heterogeneous population with different populations expressing certain markers differentially. While α-SMA myofibroblasts represent the majority of CAFs there are other markers that have been used to detect subtypes of fibroblasts [9]. However, it is unclear whether these subtypes truly represent distinct sub-populations of fibroblasts. Studies have suggested that ROS can play a role in impacting fibroblast subtype [9]. One subtype of fibroblast that may be affected by ROS are platelet-derived growth factor beta (PDGF-β) fibroblasts. ROS play an important role in PDGF signaling through the inhibition of phosphatases [64,65,66,67][64][65][66][67]. The activation of PDGF signaling stimulates fibroblast growth and motility. Interestingly, a study demonstrated that upon PDGF stimulation of normal human fibroblasts, NOX4 and DUOX4, two enzymes responsible for increasing levels of ROS within cells, modulate cell cycle entry [68]. Together these studies indicate that in PDGF-β fibroblasts ROS could play an integral role in affecting fibroblast proliferation and migration. Another fibroblast marker that could potentially be affected by ROS is Caveolin-1 (CAV-1). Studies demonstrate that when fibroblasts and tumor epithelial cells are co-cultured in the presence of oxidative stress, CAV-1 is degraded in fibroblasts which can be prevented by the treatment of antioxidant and autophagy inhibitors [69,70,71][69][70][71]. Taken together, these studies suggest that ROS produced by fibroblasts play an important role in CAF activation and differentiation. However, ROS and other metabolic byproducts are also produced in large quantities by tumor cells and could also play a role in CAF function. ROS produced by CAFs, as mentioned previously, could also augment tumorigenesis.
H2O2 is produced from tumor epithelial cells and can diffuse into other tissues and cells. H2O2 is also implicated in intracellular signaling pathway activation. Certain studies focused on understanding how H2O2 affects the tumor microenvironment (TME) and stroma. In these studies, breast cancer cells were co-cultured with CAFs to demonstrate the effect of tumor-generated H2O2 on CAFs. Interestingly, tumor H2O2 led to a reduction in mitochondrial function, increase in glucose uptake and increase ROS in CAFs [70,72][70][72]. Furthermore, co-cultured cancer cells demonstrated increased mitochondrial activity and decreased GLUT1 expression along with decreased glucose uptake [70]. This cross-talk between tumor cells and CAFs could be abrogated with the addition of catalase [70]. Finally, fibroblasts co-cultured with breast cancer cells displayed Caveolin-1 (CAV-1) downregulation and increased expression of markers for myofibroblasts [70]. This suggests that tumor cells produce ROS which can directly reprogram CAFs to potentially create a more pro-tumorigenic microenvironment.
Caveolins, such as CAV-1, are unique proteins which are found on multiple cell types and help to form caveolae which are plasma membrane invaginations. CAV-1 expression is mediated by self-digestion or autophagy [73,74][73][74]. Human CAFs will usually display reduced CAV-1 expression as compared to normal fibroblasts. Reduced CAV-1 expression is associated with increased glycolysis and reduced mitochondrial function and this decrease in CAV-1 expression is thought to be mediated by tumor cell oxidative stress induced autophagy [8,74,75][8][74][75]. Fibroblast-mediated degradation of CAV1 can be abrogated with antioxidants and autophagy inhibitors [70,71][70][71]. CAV-1 expression has not only been implicated in the induction of a metabolic switch but also in autophagy/mitophagy activity and remodeling of the microenvironment [9]. Interestingly, CAV-1 expression in lung cancer cells is differentially affected by different types of ROS. For example, hydroxyl radical up-regulates CAV-1 while O2• and H2O2 down-regulated CAV-1 expression. It should also be noted that degradation of CAV-1 leads to increased exosomal uptake into cells [76].
Exosomes are a subtype of extracellular vesicles (EVs) deriving from intraluminal endosomal vesicles. Exosomes are made up of a lipid bilayer and contain proteins, mRNAs, lipids, miRNAs and free metabolites which are released into the cytosol of target cells after internalization [77]. Cancer-derived EVs are able to transform non-malignant cells in the tumor microenvironment in order to promote tumorigenesis [78,79][78][79]. EVs, therefore, provide a mechanism for cellular crosstalk. Indeed, studies show that exosomes have the ability to reprogram recipient cells and are able to modulate proliferation, survival and immune effector status in recipient cells [80]. More recently, Zhao et al., demonstrated that exosomes isolated from prostate and pancreatic cancer patient-derived CAFs can inhibit mitochondrial oxidative phosphorylation in cancer cells increasing glycolysis and reductive carboxylation [81].
Exosomes are taken up into cells through different pathways and the process by which exosomes are taken up into cells is controversial. Recently, a study demonstrated that exosomes derived from glioblastoma (GBM) cells are internalized through non-classical, lipid-raft dependent endocytosis [76]. The authors then demonstrate that the lipid raft associated protein, CAV-1, negatively regulates the uptake of exosomes [76]. Previously, it was mentioned that tumor cells induce oxidative stress which leads to the autophagic degradation of CAV-1 [8,74][8][74]. Together, these studies suggest a pathway by which tumor cells produce ROS which signal to fibroblasts and lead to the degradation of CAV-1 and, therefore, increased exosomal uptake. The effect of increased exosomal uptake could then lead to increased influx of metabolites and metabolic reprogramming of the fibroblast to a more pro-tumorigenic CAF (i.e., myofibroblast) (Figure 2A). Indeed, Zhao et al. demonstrated that exosomes isolated from pancreatic and prostate cancer CAFs contained high amounts of glutamine, lactate and acetate as well as many other amino acids and metabolites suggesting a role for exosomes in anapleurosis and lipogenesis [81].
Autophagy is a pathway by which cytoplasmic organelles or components are sequestered into an autophagosome and delivered to lysosomes for degradation. Autophagy is essential for survival, differentiation, development and metabolism and is involved in many disease states, such as cancer. Autophagy can be stimulated by cellular stress including ROS [82,83][82][83]. ROS are able to regulate autophagy both directly and indirectly [84,85,86][84][85][86]. ROS-induced autophagy has been demonstrated to protect against oxidative damage suggesting an ROS-dependent negative feedback loop to regulate oxidative stress within cells [87]. Defective autophagy is observed in multiple tumors which supports a tumor suppressive role [88,89,90][88][89][90]. However, studies also show that autophagy has tumor-promoting functions which implies autophagy function is context dependent in cancer [91,92][91][92]. Aside from cancer type, this context-dependent functioning likely applies to cells present in the TME as well. There is now evidence that ROS can provide cross-talk between CAFs and tumor cells through autophagy that can create a more pro-tumorigenic environment. A previous study performed in a xenograft model of breast cancer demonstrated that HIF-1α-dependent activation of autophagy in stromal cells enhances tumorigenicity [93]. Given the effect of mitochondrial ROS on HIF-1α, it can be surmised that ROS in stromal cells may modulate tumorigenicity of cancer cells through induction of autophagy. Another study explored CAFs isolated from ovarian cancer tissues as well as normal fibroblasts from benign tissue and found that CAFs are resistant to oxidative stress and this process is mediated through autophagy [94]. CAFs could act as central mediators of oxidative stress within the TME and help to give tumor cells the ability to circumvent the cytotoxic effects of elevated TME ROS levels. Tumor cells can also affect the cells of TME through autophagy and mitophagy, which is the selective degradation of mitochondria by autophagy. Several studies demonstrated that tumor cells can induce increased metabolism in CAFs which also induces autophagy and mitophagy allowing for the recycling of important biomolecules and metabolic precursors [95,96][95][96]. It would be expected that the byproduct of this would also be generation of ROS in CAFs which could also help reprogram them to a more pro-tumorigenic fibroblast.
References
- Rhee, S.G. Cell signaling. H2O2 a necessary evil for cell signaling. Science 2006, 312, 1882–1883.
- Diebold, L.; Chandel, N.S. Mitochondrial ROS regulation of proliferating cells. Free Radic. Biol. Med. 2016, 100, 86–93.
- Weinberg, F.; Chandel, N.S. Reactive oxygen species-dependent signaling regulates cancer. Cell Mol. Life Sci. 2009, 66, 3663–3673.
- Weinberg, F.; Chandel, N.S. Mitochondrial metabolism and cancer. Ann. N. Y. Acad. Sci. 2009, 1177, 66–73.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793.
- Kong, H.; Chandel, N.S. Regulation of redox balance in cancer and T cells. J. Biol. Chem. 2018, 293, 7499–7507.
- Martinez-Outschoorn, U.E.; Balliet, R.M.; Rivadeneira, D.B.; Chiavarina, B.; Pavlides, S.; Wang, C.; Whitaker-Menezes, D.; Daumer, K.M.; Lin, Z.; Witkiewicz, A.K.; et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle 2010, 9, 3256–3276.
- Costa, A.; Scholer-Dahirel, A.; Mechta-Grigoriou, F. The role of reactive oxygen species and metabolism on cancer cells and their microenvironment. Semin. Cancer Biol. 2014, 25, 23–32.
- Storz, P. Reactive oxygen species in tumor progression. Front. Biosci. 2005, 10, 1881–1896.
- Szatrowski, T.P.; Nathan, C.F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991, 51, 794–798.
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13.
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013, 1, 304–312.
- Nickel, A.; Kohlhaas, M.; Maack, C. Mitochondrial reactive oxygen species production and elimination. J. Mol. Cell. Cardiol. 2014, 73, 26–33.
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344.
- Muller, F.L.; Liu, Y.; Van Remmen, H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem. 2004, 279, 49064–49073.
- Fridovich, I. Superoxide anion radical (O2-), superoxide dismutases, and related matters. J. Biol. Chem. 1997, 272, 18515–18517.
- Han, D.; Antunes, F.; Canali, R.; Rettori, D.; Cadenas, E. Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J. Biol Chem. 2003, 278, 5557–5563.
- Orr, A.L.; Vargas, L.; Turk, C.N.; Baaten, J.E.; Matzen, J.T.; Dardov, V.J.; Attle, S.J.; Li, J.; Quackenbush, D.C.; Goncalves, R.L.; et al. Suppressors of superoxide production from mitochondrial complex III. Nat. Chem. Biol. 2015, 11, 834–836.
- Winterbourn, C.C. The biological chemistry of hydrogen peroxide. Methods Enzymol. 2013, 528, 3–25.
- Echtay, K.S.; Murphy, M.P.; Smith, R.A.; Talbot, D.A.; Brand, M.D. Superoxide activates mitochondrial uncoupling protein 2 from the matrix side. Studies using targeted antioxidants. J. Biol. Chem. 2002, 277, 47129–47135.
- Frederick, R.L.; Shaw, J.M. Moving mitochondria: Establishing distribution of an essential organelle. Traffic 2007, 8, 1668–1675.
- Al-Mehdi, A.B.; Pastukh, V.M.; Swiger, B.M.; Reed, D.J.; Patel, M.R.; Bardwell, G.C.; Pastukh, V.V.; Alexeyev, M.F.; Gillespie, M.N. Perinuclear mitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Sci. Signal 2012, 5, ra47.
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514.
- Martinez-Reyes, I.; Diebold, L.P.; Kong, H.; Schieber, M.; Huang, H.; Hensley, C.T.; Mehta, M.M.; Wang, T.; Santos, J.H.; Woychik, R.; et al. TCA Cycle and Mitochondrial Membrane Potential Are Necessary for Diverse Biological Functions. Mol. Cell 2016, 61, 199–209.
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720.
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138.
- Pugh, C.W.; Ratcliffe, P.J. Regulation of angiogenesis by hypoxia: Role of the HIF system. Nat. Med. 2003, 9, 677–684.
- De Smet, F.; Segura, I.; De Bock, K.; Hohensinner, P.J.; Carmeliet, P. Mechanisms of vessel branching: Filopodia on endothelial tip cells lead the way. Arter. Thromb. Vasc. Biol. 2009, 29, 639–649.
- Ferrara, N. VEGF-A: A critical regulator of blood vessel growth. Eur. Cytokine Netw. 2009, 20, 158–163.
- Hoeben, A.; Landuyt, B.; Highley, M.S.; Wildiers, H.; Van Oosterom, A.T.; De Bruijn, E.A. Vascular endothelial growth factor and angiogenesis. Pharmacol. Rev. 2004, 56, 549–580.
- Sena, L.A.; Li, S.; Jairaman, A.; Prakriya, M.; Ezponda, T.; Hildeman, D.A.; Wang, C.R.; Schumacker, P.T.; Licht, J.D.; Perlman, H.; et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 2013, 38, 225–236.
- Gill, T.; Levine, A.D. Mitochondria-derived hydrogen peroxide selectively enhances T cell receptor-initiated signal transduction. J. Biol. Chem. 2013, 288, 26246–26255.
- Ames, B.N.; Shigenaga, M.K.; Hagen, T.M. Oxidants, antioxidants, and the degenerative diseases of aging. Proc. Natl. Acad. Sci. USA 1993, 90, 7915–7922.
- Schafer, Z.T.; Grassian, A.R.; Song, L.; Jiang, Z.; Gerhart-Hines, Z.; Irie, H.Y.; Gao, S.; Puigserver, P.; Brugge, J.S. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 2009, 461, 109–113.
- Irani, K.; Xia, Y.; Zweier, J.L.; Sollott, S.J.; Der, C.J.; Fearon, E.R.; Sundaresan, M.; Finkel, T.; Goldschmidt-Clermont, P.J. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science 1997, 275, 1649–1652.
- Chatterjee, A.; Mambo, E.; Sidransky, D. Mitochondrial DNA mutations in human cancer. Oncogene 2006, 25, 4663–4674.
- Park, J.S.; Sharma, L.K.; Li, H.; Xiang, R.; Holstein, D.; Wu, J.; Lechleiter, J.; Naylor, S.L.; Deng, J.J.; Lu, J.; et al. A heteroplasmic, not homoplasmic, mitochondrial DNA mutation promotes tumorigenesis via alteration in reactive oxygen species generation and apoptosis. Hum. Mol. Genet. 2009, 18, 1578–1589.
- Sharma, L.K.; Fang, H.; Liu, J.; Vartak, R.; Deng, J.; Bai, Y. Mitochondrial respiratory complex I dysfunction promotes tumorigenesis through ROS alteration and AKT activation. Hum. Mol. Genet. 2011, 20, 4605–4616.
- Woo, D.K.; Green, P.D.; Santos, J.H.; D’Souza, A.D.; Walther, Z.; Martin, W.D.; Christian, B.E.; Chandel, N.S.; Shadel, G.S. Mitochondrial genome instability and ROS enhance intestinal tumorigenesis in APC(Min/+) mice. Am. J. Pathol. 2012, 180, 24–31.
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348.
- Ostman, A.; Augsten, M. Cancer-associated fibroblasts and tumor growth--bystanders turning into key players. Curr. Opin. Genet. Dev. 2009, 19, 67–73.
- Serini, G.; Gabbiani, G. Mechanisms of myofibroblast activity and phenotypic modulation. Exp. Cell Res. 1999, 250, 273–283.
- Madar, S.; Goldstein, I.; Rotter, V. ‘Cancer associated fibroblasts’—More than meets the eye. Trends Mol. Med. 2013, 19, 447–453.
- Madar, S.; Brosh, R.; Buganim, Y.; Ezra, O.; Goldstein, I.; Solomon, H.; Kogan, I.; Goldfinger, N.; Klocker, H.; Rotter, V. Modulated expression of WFDC1 during carcinogenesis and cellular senescence. Carcinogenesis 2009, 30, 20–27.
- Buganim, Y.; Madar, S.; Rais, Y.; Pomeraniec, L.; Harel, E.; Solomon, H.; Kalo, E.; Goldstein, I.; Brosh, R.; Haimov, O.; et al. Transcriptional activity of ATF3 in the stromal compartment of tumors promotes cancer progression. Carcinogenesis 2011, 32, 1749–1757.
- Rasmussen, A.A.; Cullen, K.J. Paracrine/autocrine regulation of breast cancer by the insulin-like growth factors. Breast Cancer Res. Treat. 1998, 47, 219–233.
- Noel, A.; De Pauw-Gillet, M.C.; Purnell, G.; Nusgens, B.; Lapiere, C.M.; Foidart, J.M. Enhancement of tumorigenicity of human breast adenocarcinoma cells in nude mice by matrigel and fibroblasts. Br. J. Cancer 1993, 68, 909–915.
- Kuperwasser, C.; Chavarria, T.; Wu, M.; Magrane, G.; Gray, J.W.; Carey, L.; Richardson, A.; Weinberg, R.A. Reconstruction of functionally normal and malignant human breast tissues in mice. Proc. Natl. Acad. Sci. USA 2004, 101, 4966–4971.
- Gascard, P.; Tlsty, T.D. Carcinoma-associated fibroblasts: Orchestrating the composition of malignancy. Genes Dev. 2016, 30, 1002–1019.
- Chauhan, V.P.; Boucher, Y.; Ferrone, C.R.; Roberge, S.; Martin, J.D.; Stylianopoulos, T.; Bardeesy, N.; DePinho, R.A.; Padera, T.P.; Munn, L.L.; et al. Compression of pancreatic tumor blood vessels by hyaluronan is caused by solid stress and not interstitial fluid pressure. Cancer Cell 2014, 26, 14–15.
- Jezierska-Drutel, A.; Rosenzweig, S.A.; Neumann, C.A. Role of oxidative stress and the microenvironment in breast cancer development and progression. Adv. Cancer Res. 2013, 119, 107–125.
- Radisky, E.S.; Radisky, D.C. Stromal induction of breast cancer: Inflammation and invasion. Rev. Endocr. Metab. Disord. 2007, 8, 279–287.
- Zavadil, J.; Haley, J.; Kalluri, R.; Muthuswamy, S.K.; Thompson, E. Epithelial-mesenchymal transition. Cancer Res. 2008, 68, 9574–9577.
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007, 67, 10123–10128.
- McDonald, L.T.; Russell, D.L.; Kelly, R.R.; Xiong, Y.; Motamarry, A.; Patel, R.K.; Jones, J.A.; Watson, P.M.; Turner, D.P.; Watson, D.K.; et al. Hematopoietic stem cell-derived cancer-associated fibroblasts are novel contributors to the pro-tumorigenic microenvironment. Neoplasia 2015, 17, 434–448.
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011, 71, 2455–2465.
- Kojima, Y.; Acar, A.; Eaton, E.N.; Mellody, K.T.; Scheel, C.; Ben-Porath, I.; Onder, T.T.; Wang, Z.C.; Richardson, A.L.; Weinberg, R.A.; et al. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc. Natl. Acad. Sci. USA 2010, 107, 20009–20014.
- Mueller, L.; Goumas, F.A.; Affeldt, M.; Sandtner, S.; Gehling, U.M.; Brilloff, S.; Walter, J.; Karnatz, N.; Lamszus, K.; Rogiers, X.; et al. Stromal fibroblasts in colorectal liver metastases originate from resident fibroblasts and generate an inflammatory microenvironment. Am. J. Pathol. 2007, 171, 1608–1618.
- Toullec, A.; Gerald, D.; Despouy, G.; Bourachot, B.; Cardon, M.; Lefort, S.; Richardson, M.; Rigaill, G.; Parrini, M.C.; Lucchesi, C.; et al. Oxidative stress promotes myofibroblast differentiation and tumour spreading. EMBO Mol. Med. 2010, 2, 211–230.
- Jain, M.; Rivera, S.; Monclus, E.A.; Synenki, L.; Zirk, A.; Eisenbart, J.; Feghali-Bostwick, C.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial reactive oxygen species regulate transforming growth factor-beta signaling. J. Biol. Chem. 2013, 288, 770–777.
- Artaud-Macari, E.; Goven, D.; Brayer, S.; Hamimi, A.; Besnard, V.; Marchal-Somme, J.; Ali, Z.E.; Crestani, B.; Kerdine-Romer, S.; Boutten, A.; et al. Nuclear factor erythroid 2-related factor 2 nuclear translocation induces myofibroblastic dedifferentiation in idiopathic pulmonary fibrosis. Antioxid. Redox Signal. 2013, 18, 66–79.
- Sampson, N.; Koziel, R.; Zenzmaier, C.; Bubendorf, L.; Plas, E.; Jansen-Durr, P.; Berger, P. ROS signaling by NOX4 drives fibroblast-to-myofibroblast differentiation in the diseased prostatic stroma. Mol. Endocrinol. 2011, 25, 503–515.
- Frijhoff, J.; Dagnell, M.; Augsten, M.; Beltrami, E.; Giorgio, M.; Ostman, A. The mitochondrial reactive oxygen species regulator p66Shc controls PDGF-induced signaling and migration through protein tyrosine phosphatase oxidation. Free Radic. Biol. Med. 2014, 68, 268–277.
- Dagnell, M.; Frijhoff, J.; Pader, I.; Augsten, M.; Boivin, B.; Xu, J.; Mandal, P.K.; Tonks, N.K.; Hellberg, C.; Conrad, M.; et al. Selective activation of oxidized PTP1B by the thioredoxin system modulates PDGF-beta receptor tyrosine kinase signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 13398–13403.
- Meng, T.C.; Fukada, T.; Tonks, N.K. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol. Cell. 2002, 9, 387–399.
- Sundaresan, M.; Yu, Z.X.; Ferrans, V.J.; Irani, K.; Finkel, T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 1995, 270, 296–299.
- Salmeen, A.; Park, B.O.; Meyer, T. The NADPH oxidases NOX4 and DUOX2 regulate cell cycle entry via a p53-dependent pathway. Oncogene 2010, 29, 4473–4484.
- Sotgia, F.; Martinez-Outschoorn, U.E.; Howell, A.; Pestell, R.G.; Pavlides, S.; Lisanti, M.P. Caveolin-1 and cancer metabolism in the tumor microenvironment: Markers, models, and mechanisms. Annu. Rev. Pathol. 2012, 7, 423–467.
- Martinez-Outschoorn, U.E.; Pavlides, S.; Whitaker-Menezes, D.; Daumer, K.M.; Milliman, J.N.; Chiavarina, B.; Migneco, G.; Witkiewicz, A.K.; Martinez-Cantarin, M.P.; Flomenberg, N.; et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: Implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle 2010, 9, 2423–2433.
- Martinez-Outschoorn, U.E.; Whitaker-Menezes, D.; Lin, Z.; Flomenberg, N.; Howell, A.; Pestell, R.G.; Lisanti, M.P.; Sotgia, F. Cytokine production and inflammation drive autophagy in the tumor microenvironment: Role of stromal caveolin-1 as a key regulator. Cell Cycle 2011, 10, 1784–1793.
- Martinez-Outschoorn, U.E.; Lin, Z.; Trimmer, C.; Flomenberg, N.; Wang, C.; Pavlides, S.; Pestell, R.G.; Howell, A.; Sotgia, F.; Lisanti, M.P. Cancer cells metabolically “fertilize” the tumor microenvironment with hydrogen peroxide, driving the Warburg effect: Implications for PET imaging of human tumors. Cell Cycle 2011, 10, 2504–2520.
- Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Caveolae and signalling in cancer. Nat. Rev. Cancer 2015, 15, 225–237.
- Martinez-Outschoorn, U.E.; Trimmer, C.; Lin, Z.; Whitaker-Menezes, D.; Chiavarina, B.; Zhou, J.; Wang, C.; Pavlides, S.; Martinez-Cantarin, M.P.; Capozza, F.; et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival: Role of hypoxia, HIF1 induction and NFkappaB activation in the tumor stromal microenvironment. Cell Cycle 2010, 9, 3515–3533.
- Asterholm, I.W.; Mundy, D.I.; Weng, J.; Anderson, R.G.; Scherer, P.E. Altered mitochondrial function and metabolic inflexibility associated with loss of caveolin-1. Cell Metab. 2012, 15, 171–185.
- Svensson, K.J.; Christianson, H.C.; Wittrup, A.; Bourseau-Guilmain, E.; Lindqvist, E.; Svensson, L.M.; Morgelin, M.; Belting, M. Exosome uptake depends on ERK1/2-heat shock protein 27 signaling and lipid Raft-mediated endocytosis negatively regulated by caveolin-1. J. Biol. Chem. 2013, 288, 17713–17724.
- Zhao, H.; Achreja, A.; Iessi, E.; Logozzi, M.; Mizzoni, D.; Di Raimo, R.; Nagrath, D.; Fais, S. The key role of extracellular vesicles in the metastatic process. Biochim. Biophys. Acta 2017, 1869, 64–77.
- Cho, J.A.; Park, H.; Lim, E.H.; Lee, K.W. Exosomes from breast cancer cells can convert adipose tissue-derived mesenchymal stem cells into myofibroblast-like cells. Int. J. Oncol. 2012, 40, 130–138.
- Paggetti, J.; Haderk, F.; Seiffert, M.; Janji, B.; Distler, U.; Ammerlaan, W.; Kim, Y.J.; Adam, J.; Lichter, P.; Solary, E.; et al. Exosomes released by chronic lymphocytic leukemia cells induce the transition of stromal cells into cancer-associated fibroblasts. Blood 2015, 126, 1106–1117.
- Gangoda, L.; Boukouris, S.; Liem, M.; Kalra, H.; Mathivanan, S. Extracellular vesicles including exosomes are mediators of signal transduction: Are they protective or pathogenic? Proteomics 2015, 15, 260–271.
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; San Lucas, F.A.; et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife 2016, 5, e10250.
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell. 2010, 40, 280–293.
- Yang, Z.; Klionsky, D.J. Eaten alive: A history of macroautophagy. Nat. Cell Biol. 2010, 12, 814–822.
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760.
- Carroll, B.; Otten, E.G.; Manni, D.; Stefanatos, R.; Menzies, F.M.; Smith, G.R.; Jurk, D.; Kenneth, N.; Wilkinson, S.; Passos, J.F.; et al. Oxidation of SQSTM1/p62 mediates the link between redox state and protein homeostasis. Nat. Commun. 2018, 9, 256.
- Li, L.; Chen, Y.; Gibson, S.B. Starvation-induced autophagy is regulated by mitochondrial reactive oxygen species leading to AMPK activation. Cell. Signal. 2013, 25, 50–65.
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388.
- Guo, J.Y.; Karsli-Uzunbas, G.; Mathew, R.; Aisner, S.C.; Kamphorst, J.J.; Strohecker, A.M.; Chen, G.; Price, S.; Lu, W.; Teng, X.; et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013, 27, 1447–1461.
- Lan, S.H.; Wu, S.Y.; Zuchini, R.; Lin, X.Z.; Su, I.J.; Tsai, T.F.; Lin, Y.J.; Wu, C.T.; Liu, H.S. Autophagy suppresses tumorigenesis of hepatitis B virus-associated hepatocellular carcinoma through degradation of microRNA-224. Hepatology 2014, 59, 505–517.
- Liu, X.D.; Yao, J.; Tripathi, D.N.; Ding, Z.; Xu, Y.; Sun, M.; Zhang, J.; Bai, S.; German, P.; Hoang, A.; et al. Autophagy mediates HIF2alpha degradation and suppresses renal tumorigenesis. Oncogene 2015, 34, 2450–2460.
- Cianfanelli, V.; Fuoco, C.; Lorente, M.; Salazar, M.; Quondamatteo, F.; Gherardini, P.F.; De Zio, D.; Nazio, F.; Antonioli, M.; D’Orazio, M.; et al. AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat. Cell Biol. 2015, 17, 706.
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gelinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64.
- Capparelli, C.; Whitaker-Menezes, D.; Guido, C.; Balliet, R.; Pestell, T.G.; Howell, A.; Sneddon, S.; Pestell, R.G.; Martinez-Outschoorn, U.; Lisanti, M.P.; et al. CTGF drives autophagy, glycolysis and senescence in cancer-associated fibroblasts via HIF1 activation, metabolically promoting tumor growth. Cell Cycle 2012, 11, 2272–2284.
- Wang, Q.; Xue, L.; Zhang, X.; Bu, S.; Zhu, X.; Lai, D. Autophagy protects ovarian cancer-associated fibroblasts against oxidative stress. Cell Cycle 2016, 15, 1376–1385.
- Pavlides, S.; Vera, I.; Gandara, R.; Sneddon, S.; Pestell, R.G.; Mercier, I.; Martinez-Outschoorn, U.E.; Whitaker-Menezes, D.; Howell, A.; Sotgia, F.; et al. Warburg meets autophagy: Cancer-associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid. Redox Signal. 2012, 16, 1264–1284.
- Martinez-Outschoorn, U.E.; Pavlides, S.; Howell, A.; Pestell, R.G.; Tanowitz, H.B.; Sotgia, F.; Lisanti, M.P. Stromal-epithelial metabolic coupling in cancer: Integrating autophagy and metabolism in the tumor microenvironment. Int. J. Biochem. Cell Biol. 2011, 43, 1045–1051.
More