In vitro chemical antioxidant assays can generally be classified into two: those involving hydrogen atom transfer (HAT) and those based on electron transfer (ET)
[18]. While most HAT reactions such as lipid peroxidation (including lipoprotein oxidation) and oxygen radical absorbance capacity (ORAC) measure the capacity of an antioxidant to inactivate a free radical through the release of a hydrogen atom, assays based on single electron transfer such as ferric iron reducing antioxidant power (FRAP), 2,2-diphenyl-1-picrylhydrazyl (DPPH), and 2,2′-azinobis[3-ethylbenzothiazoline-6-sulfonic acid] (ABTS) measure the release or transfer of an electron to a free radical, thus converting it into an anion
[18][19][18,19]. A number of technical and conceptual impediments that preclude the use and impair the validity of in vitro antioxidant assays have been discussed
[17]. These limitations include the unsuitability of the chemistry and molecular targets of most in vitro assays to the in vivo environment, the inadequacy of commonly used antioxidant assays in measuring the radical reactions in lipids, the ambiguity of the chemistry of the assays with regard to quantitative analyses, and the near absence of standardization in the experimental procedures of many currently used antioxidant assays
[17]. With respect to the poor correlation between the chemistry and molecular targets of in vitro assays and that of the in vivo environment, for instance, it has been noted that the direct scavenging of the extremely reactive and short-lived hydroxyl radical (
•OH) by dietary antioxidants in vivo is unrealistic and of diminished physiological relevance. This is because the intracellular concentration of dietary antioxidants is negligible and thus insufficient to scavenge this most powerful biological oxidizing intermediate
[20], which has the capacity to hydroxylate biological macromolecules such as proteins, nucleic acids, and lipids
[18]. In addition, it is recognized that careful consideration must be given to the type of solvent used in antioxidant assays in order to avoid false positive results
[15], since the type of solvent used could significantly affect test results
[10]. For instance, in a study examining the effect of various solvent types viz methanol, isopropanol, chloroform, acetone, hexane, and ethyl acetate on the amount of unreacted DPPH radical, the highest concentration of DPPH radical remaining after a reaction time of 60 min was observed for ethyl acetate and the lowest was observed for chloroform
[21]. While the DPPH reagent is notable for its affinity toward hydrophobic solvents in contradistinction to its antipathy for hydrophilic ones, the ABTS reagent can effectively dissolve in both lipophilic and hydrophilic solvents
[22]. The sub-sections that follow provide a concise discussion of the various bases of chemical antioxidant assays such as scavenging of free radicals, trapping of reactive oxygen species, reduction of ferric iron to the more stable Fe
2+ form, prooxidant chelation of metal ions, and inhibition of lipid peroxidation.
2.1. Free Radical (Synthetic DPPH) Scavenging
In living cells, a natural antioxidant defense system consisting of endogenous enzymatic antioxidants exists to counteract the actions of ROS such as superoxide anion and hydrogen peroxide, while a variety of non-enzymatic antioxidants such as ascorbic acid and α-tocopherols are tasked with the responsibility of scavenging free radicals and oxidants such as peroxynitrite, hydroxyl radical, singlet oxygen, and peroxyl radicals
[10][18][23][10,18,23]. Since they are rapid, simple, relatively straightforward, and largely inexpensive, a number of antioxidant assays designed to measure the capacity of antioxidants to scavenge free radicals are widely used in food and biological science research to evaluate the free radical/oxidant-scavenging property of biological and food samples
[15][18][23][15,18,23].
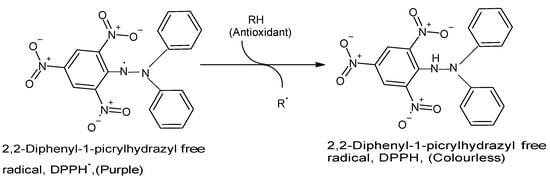
The DPPH assay has become one of the most commonly used in vitro chemical antioxidant tests mainly because it is highly sensitive, technically simple, rapid, accurate, reproducible, reliable, and does not require any special sample pre-treatment
[10][15][23][24][25][10,15,23,24,25]. The assay is usually performed by combining a methanolic DPPH solution (25 mg/L) with the test sample solution and monitoring the absorbance of the mixture at 515–517 nm using a spectrophotometer for 30 min or until the absorbance is stable
[15][18][15,18]. A strong (purple) absorption maximum is shown by the DPPH organic nitrogen radical at 517 nm, and the antioxidant capacity of the test sample is directly proportional to the disappearance of color, i.e., reduced absorbance
[15][23][15,23]. Upon reduction of the DPPH radical by and following hydrogen atom abstraction from the antioxidant, the solution loses its color and fades from purple to pale yellow
[15][23][15,23] (
Figure 1). Then, absorbance (A
s) of the reaction containing the antioxidant molecule is subtracted from absorbance (A
b) of the reaction without the sample (blank) and expressed as percentage loss. When the reaction is performed at different sample (antioxidant) concentrations, a plot of inhibition rate versus concentration allows for calculation of the EC
50 (effective concentration of the antioxidant needed to reduce the amount of DPPH radicals by half)
[17].
Figure 1. Reaction between DPPH radical and an antioxidant to yield the colorless DPPH. The reaction of DPPH radical with other radicals, hydrogen atoms, or electrons results in the loss of color at 515 nm. (Chemical structures produced with ACD/ChemSketch Freeware Version 2021.1.0 C25E41).
In contrast to most other free radicals, the DPPH molecule does not undergo dimerization because the spare electron is delocalized over the entire molecule, resulting in the formation of a deep violet color in aqueous, methanolic, and ethanolic solutions in which the radical has been shown to rarely disintegrate
[25]. Since DPPH is hydrophobic, assays involving the free radical must be performed in organic solvents
[17]. The characteristics of the selected solvent and pH can influence DPPH scavenging activity, thus highlighting the importance of carefully considering solvent property in order to avoid false positive results
[15]. It was reported that in the reaction between DPPH and phenols, the rate-determining step is the very rapid transfer of electron from the phenoxide anions to DPPH, while the hydrogen atom transfer from the neutral phenol to DPPH represents a marginal reaction step, since it occurs at a much slower rate in strong hydrogen bond-accepting solvents such as alcohol and methyl alcohol
[26]. In a study of crude palm oil-derived carotenoids, the finding that α- and β-carotenes were better DPPH radical scavengers than metal chelators was suggested to be probably due to the presence of unsaturated groups in the tetraterpenoids
[27]. Limitations of the DPPH assay include that it misses critical data present in reaction curves, since it does not measure reaction rates as well as the interaction of DPPH with dissolved oxygen, which could be an issue for compounds that autoxidize such as certain phenols and ascorbate
[17]. In addition, the steric inaccessibility of the DPPH radical site, which impairs its interaction with samples such as fruits and vegetables extracts that typically contain a mixture of antioxidants also limits the reliability of the test
[17]. For instance, samples containing eugenol and similar phenols with the o-methoxyphenol structure are known to give falsely low antioxidant capacity readings
[18]. Results from the use of DPPH as a colorimetric probe for the detection of free radical scavengers are often reported as EC
50 or as TEC
50, which is defined as the time required to reach steady state with EC
50 [10]. However, the usefulness of interpreting DPPH assay data in this manner has been called into question, since EC
50 is time-dependent, and the effect of time is not uniform for all compounds, and also because EC
50 is a concentration, not a kinetic parameter, and therefore cannot accurately be used to denote the antioxidant or antiradical capacity of a compound
[28]. Moreover, the DPPH radical is a synthetic compound that is not present in plant and animal tissues; therefore, the data obtained may not reflect the true radical scavenging ability of a molecule within the human body or as food preservatives. Hence, extrapolation of the DPPH radical scavenging data to the antioxidant capacity of a molecule in real life or food product situations must be done with caution.
2.2. ROS Trapping (Hydrogen Peroxide, Nitric Oxide, Also Superoxide and Hydroxyl)
Of the six major ROS (superoxide anion, hydrogen peroxide, peroxyl radicals, hydroxyl radical, singlet oxygen, and peroxynitrite) known to cause oxidative damage in the human body, two endogenous antioxidant enzymes, namely superoxide dismutase and catalase, are known to neutralize superoxide anion and hydrogen peroxide, respectively
[18][26][18,26]. In contrast, non-enzymatic antioxidants such as phytochemicals, ascorbic acid, and alpha-tocopherol are saddled with the responsibility of scavenging the other four oxidants and free radicals
[18][29][18,29]. Various in vitro ROS trapping assays exist for evaluating the oxidant scavenging capacity of biological samples. For instance, the hydroxyl radical scavenging activity of a range of biological samples such as chicken skin enzymatic protein hydrolysates
[30] and peach fruit extracts
[31] has been evaluated using a method proposed by de Avellar et al.
[32]. This method is based on the production of hydroxyl radicals in the Fenton reaction following the combination of hydrogen peroxide with 1,10-phenanthroline and ferrous ammonium sulfate. Although hydrogen peroxide is chemically unreactive at low concentrations, under physiological conditions, its oxidation power can be observed in combination with ferrous ion in the Fenton reaction
[18]. With regard to the unsuitability of the chemistry and molecular targets of in vitro assays to in vivo reactions, for instance, it has been noted that the Fe
2+/H
2O
2 mixture used in scavenging assays has a certain drawback, since many antioxidants are also metal chelators
[18]. Thus, mixing the antioxidant sample with Fe
2+ could alter the activity of the ferrous ion by chelation, thus making it impossible to determine if the antioxidant is an effective hydroxyl radical scavenger or just a good metal chelator
[18].
2.3. Ferric Reducing Antioxidant Power (FRAP)
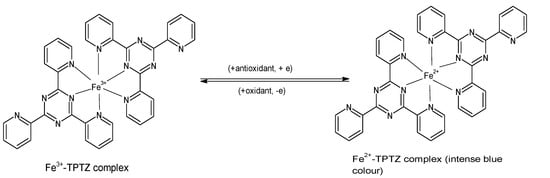
The FRAP assay evaluates the ability of antioxidants to reduce ferric iron in the form of Fe
3+-2,4,6-tripyridyl-S-triazine (TPTZ) complex to the more stable, divalent Fe
2+ ion at low pH
[8][33][8,33]. Initially developed to determine the concentration of ascorbic acid in plasma
[34], the assay measures the change in absorbance using a spectrophotometer, and it is performed by incubating 300 µL of freshly prepared “FRAP reagent” (25 mL acetate buffer, 2.5 mL FeCl
3·H
2O (20 mM), and 2.5 mL of 10 mM TPTZ in 40 mM HCl) with a reagent blank at 37 °C for 30 min, and taking a reading at 593 nm. Subsequently, the test sample (10 µL) and water (30 µL) are added to the reaction mixture, and absorbance readings are taken after 0.5 s and thereafter, every 15 s for 4 min
[15][18][15,18]. The reduction to Fe
2+ that yields a violet-blue color (
Figure 2) provides a quick, reproducible result and has been used in many studies for determining the antioxidant capacity of various foods including vegetables, cereals, fruits, beans, and essential oils
[23][35][36][37][38][39][23,35,36,37,38,39]. Certain polyphenols such as ascorbic acid, quercetin, ferulic acid, caffeic acid, and tannic acid have been reported to show increasing absorbance (A
593) beyond the standard assay reaction time of 4 min, thus the higher FRAP values of such compounds
[40].
Figure 2. Reaction of ferric tripyridyl-S-triazine complex with antioxidant to yield the intensely blue ferrous form of the complex at an absorbance maxima of 593 nm. (Chemical structures produced with ACD/ChemSketch Freeware Version 2021.1.0 C25E41).
In food protein-derived bioactive peptides, certain properties such as a terminal methionine residue, the presence of sulfur-containing amino acids, and amino acid hydrophobicity have been reported to enhance FRAP. In contrast, the presence of lysine residues and a high content of cationic amino acids in protein hydrolysates is thought to impair their FRAP potential
[3][8][41][42][3,8,41,42]. In a study that investigated various antioxidant activities of 13 apple cultivars, the highest FRAP values were recorded for the apple peel extracts, which contain higher phenolics and flavonoids, and lower ascorbic acid levels compared to the samples from the apple cortex
[35]. The ferric reducing capacity (FRC) assay, which is similar to the FRAP assay in many aspects including in lacking the capacity to measure thiol groups, but which differs from the latter in replacing TPTZ with 1,10-phenanthroline (since phenanthroline forms a Fe
3+-[Phen]
3 complex that is reduced to an orange-red Fe
2+-(Phen) complex), has been used as an alternative to FRAP due to its simplicity and speed
[9]. In a study comparing the performance of both assays in measuring the total antioxidant capacity (TAC) of serum samples, serum TAC values ranged from 172 to 418 μmol/L of vitamin C equivalents for FRAP and from 264 to 610 for FRC, with a Spearman rank order correlation result (rs = 0.75,
p = 0.01) suggesting a strong positive correlation between the TAC of the serum samples measured by both assay methods
[9]. Another study compared the antioxidant activities of medicinal plant infusions using a modified FRC assay (the highly ferrous-stabilizing ligand ferrozine) and the conventional FRAP test
[43]. It was found that the FRC was superior to FRAP with respect to faster kinetics, enhanced sensitivity, and absence of free Fe (II), which has been shown to lead to Fenton-type oxidations in reaction products. In the modified FRC assay, ferric ion, in the presence of ferrozine, is reduced to the magenta colored Fe
2+-ferrozine complex with absorption maxima at 562 nm
[43]. A more recent modification of the FRAP assay in which the spectrophotometric quantification of the Prussian blue end product is used to determine antioxidant reducing power involves the use of potassium ferricyanide
[10]. In this iteration of the FRAP assay, the antioxidant sample either reduces the ferric ion in the solution to ferrous ion, which then complexes with the ferricyanide to yield Prussian blue or reduces the ferricyanide to ferrocyanide that interacts with the free ferric ion in the solution to form Prussian blue
[10][44][10,44].
Certain limitations impair the validity, efficiency, usefulness, and accuracy of the FRAP assay. For instance, it must be performed in an aqueous system, thus necessitating the use of a water-soluble reference antioxidant such as Trolox, ascorbic acid, or uric acid
[23]. Since the oxidant in the “FRAP reagent” is not only Fe
3+[TPTZ]
2 but also other ferric species, evaluating the antioxidative potential of foods using this assay could be problematic because many metal chelators in food extracts are able to bind to Fe
3+, forming complexes that have the capacity to react with antioxidants
[18]. In addition, the assay is notorious for its inability to accurately measure the antioxidant activity of slow-reacting compounds such as polyphenols and for giving false-positive results for samples with redox potential values lower than the Fe
3+/Fe
2+ redox pair
[15]. It has been noted that at approximately 0.70 V, there is little difference between the redox potential of the Fe
3+ salt and that of the ABTS radical at 0.68 V, thus making differential pH (neutral for Trolox equivalent antioxidant capacity and acidic for FRAP) one of the only real differences between the two assays
[18]. In the ferricyanide FRAP assay, it has been noted that the Prussian blue end product often precipitates to form a suspension, which contributes to staining the test cuvette and raising the potential for error in the assay
[10].
2.4. Prooxidant Metal Chelation
Metal ions possess the capacity to induce lipid oxidation by means of the Fenton reaction as well as by breaking down lipid hydroperoxides into more reactive radicals
[10]. Certain natural antioxidants including flavonoids such as quercetin, rutin, and (+)catechin have been shown to be powerful metal chelators
[10][45][10,45]. Metal chelators function as antioxidants by scavenging ROS and also by decreasing the amount of available metals such as iron, thereby reducing the amount of hydroxyl radicals generated by Fenton reactions and limiting metal ion-induced lipid oxidation
[10][46][10,46]. Since the antioxidant capacity of metal ion chelators is determined when a complex is formed between the antioxidant and the metal, which renders the metal ion unavailable to function as an initiator of lipid oxidation, metal chelation capacity is used as an indicator of antioxidant activity
[10]. A common assay for investigating the capacity of biological samples to chelate metal ions involves the combination of FeCl
2, ferrozine, and the test biological sample. Then, the absorbance readings at 562 nm is used as a measure of the metal ion-chelating capacity of the antioxidant sample
[11][47][11,47]. Another adaptation of the assay uses ferrous sulfate in place of ferrozine and measures absorbance at 485 nm
[10]. An important investigation of the metal ion-chelating property of various natural flavonoids found that flavonoids can act as both prooxidants and antioxidants depending on the nature and concentration of the flavonoids and metal ions
[45].
2.5. Inhibition of Lipid Peroxidation including Lipoprotein Oxidation
Lipid oxidation is a major contributor to the impairment of food quality and industrial-scale economic losses
[8]. In biological systems, lipid peroxidation greatly contributes to oxidative damage in cell membranes, lipoproteins, and other lipid-containing structures
[48]. Iron-catalyzed one-electron reduction of lipid hydroperoxides can lead to free radical-mediated chain peroxidation and the perturbation of cell membrane structure and function and associated pathologies, while peroxyl radicals are known to play a vital role in the unsavory oxidation of lipids in food and biological systems
[18][48][18,48]. In addition, when exposed to heat, light, enzymes, metalloproteins and metals, lipids are subject to oxidative processes, which could result in the development of rancidity and off-flavors and the consequent loss of essential organoleptic and nutritional qualities
[8][10][8,10]. Lipid oxidation could be a result of autoxidation, photooxidation, thermal oxidation, or enzymatic oxidation
[10]. The inhibition of linoleic acid oxidation by food extracts has been used to study lipid oxidation in vitro
[8]. The assay typically involves the combination of an ethanolic linoleic acid solution with the antioxidant test sample and incubation at 60 °C in the dark for seven days
[49]. Measurements are taken every 24 h for the entire duration of the incubation and typically entails aspirating and combining a specific amount of the previously incubated mixture with aqueous ethanol, ammonium thiocyanate, and FeCl
2 and taking spectrophotometric measurements at 500 nm at room temperature
[49]. Reductions in the absorbance value of samples when compared to blank reaction (containing no antioxidant molecules) are used as a measure of the inhibitory potency of the antioxidant compound against lipid peroxidation.