Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Barbara Stefanska and Version 2 by Vivi Li.
Epigenetic aberrations are linked to sporadic breast cancer. Interestingly, certain dietary polyphenols with anti-cancer effects, such as pterostilbene (PTS), have been shown to regulate gene expression by altering epigenetic patterns. Our group has proposed the involvement of DNA methylation and DNA methyltransferase 3B (DNMT3B) as vital players in PTS-mediated suppression of candidate oncogenes and suggested a role of enhancers as target regions.
- pterostilbene
- enhancer
- DNA methylation
- DNMT3B
- H3K36 trimethylation
- oncogenes
- chromatin
- cancer
1. Introduction
Breast cancer is the most commonly diagnosed malignancy and the second leading cause of cancer-related deaths among women [1]. Epigenetic alterations are found frequently in breast cancer and are suggested to drive a number of sporadic cases [2]. Epigenetics refers to the control of gene expression without changes to the DNA sequence. The epigenetic code consists of DNA methylation, covalent histone modifications, noncoding RNA mechanisms, and chromatin remodeling complexes [3][4][3,4]. All components of the epigenetic machinery are important to influence gene expression; however, DNA methylation mainly occurs on the cytosine of CpG dinucleotides and is catalyzed by specific enzymes, namely, DNA methyltransferases (DNMTs), and they are particularly considered to stabilize gene transcription [3][4][5][3,4,5]. DNA methylation reactions are thought to provide stable, long-term regulation of gene expression over time [6], while at the same time are considered to be dynamic and responsive to external exposures [7]. During carcinogenesis, perturbations in the DNA methylation machinery occur, leading to aberrant DNA methylation patterns in sequences that are involved in the regulation of gene transcription, which subsequently affects the expression of genes related to cell differentiation, proliferation, metastasis, and other biological processes [8]. For decades, researchers were reporting on aberrant DNA methylation levels within promoters of genes while the role and regulation of other regulatory regions, e.g., enhancers, silencers, or insulators, were largely disregarded [9]. Studies over the last 20 years clearly demonstrate that the DNA methylation status of enhancer regions may impact their regulatory role in addition to histone marks [10].
Enhancers are usually 30–200 base pairs long and may be located upstream or downstream of their target genes [9][11][9,11]. The activity of enhancers is highly dependent on a cell type, physiological stimuli, and a specific developmental time point. These distant regulatory elements increase the likelihood of gene transcription by interacting with their target gene promoters via looping of DNA in association with transcription factors, RNA polymerase, and other cofactors [9][11][9,11]. Another way through which enhancers can impact the expression of target genes is through their own transcription and generating enhancer RNA (eRNA), which positively correlates with mRNA synthesis of the target genes [12]. Several classes of enhancers have been described, including active, intermediate, and poised, that are characterized by distinct histone marks [13]. For example, active enhancers are rich in acetylation of lysine 27 of histone H3 (H3K27ac), whereas poised enhancers are marked by trimethylation of H3K27 (H3K27me3) [9][13][9,13]. Of importance, H3K36me3 has been shown as a unique mark of active enhancers, distinguishing active from intermediate or poised enhancers [13]. It is crucial to emphasize that H3K36me3 is a mark generally associated with actively transcribed genes and transcriptional elongation; thereby, it is also present at other regions relevant to transcription [13]. Apart from distinct patterns of histone covalent modifications, active enhancers display less DNA methylation [10][14][10,14]. DNA hypomethylation was also proposed as a signature of eRNA-generating enhancers, thus active enhancers [15]. While our knowledge about the regulation of enhancers is constantly increasing, approaches to reverse aberrant enhancer activity in disease remain undiscovered.
Remarkably, the last decade of research has shown epigenetic activities exerted by compounds with polyphenolic structure, including stilbenoid polyphenols [16][17][18][19][20][21][22][16,17,18,19,20,21,22], accompanied by their potent anti-cancer effects [23][24][25][23,24,25]. Studies have shown loci-specific changes in DNA methylation and reversal of cancer-like DNA methylation patterns [16][17][18][19][20][21][16,17,18,19,20,21]. Among these compounds, Pterostilbene (PTS), a naturally present stilbenoid in the diet (e.g., blueberries, grapes, cranberries, peanuts, pistachios, cocoa), stands out due to its high bioavailability [26][27][26,27]. We and others previously observed changes in DNA methylation at candidate gene loci, including PTEN, APC, and BRCA1 in human cancer cells exposed to PTS or its analog resveratrol (RSV) [16][17][18][16,17,18]. These reports were strengthened by our genome-wide study to demonstrate remodeling of the DNA methylation patterns in breast cancer cells exposed to stilbenoids [20]. We observed increases and decreases in DNA methylation levels at thousands of CpG loci. These findings laid the framework for subsequent work to identify mechanisms underlying stilbenoid-mediated changes in loci-specific DNA methylation [20][28][20,28]. For the first time, we demonstrated that stilbenoid-mediated downregulation of an oncogene, MAML2, which is an activator of the NOTCH signaling pathway, is accompanied by increased DNA methylation at the MAML2 enhancer region [20]. DNMT3B was found to bind the MAML2 enhancer upon treatment with stilbenoids, suggesting a role for this DNA methyltransferase in methylating the enhancer [20]. We further showed that one-third of hypermethylation events in response to stilbenoids occur in gene bodies, and the majority of those loci within gene bodies are located in gene enhancers [20]. In the current study, we have therefore investigated how stilbenoids, specifically PTS, impact epigenetic marks at enhancer regions in MCF10CA1a breast cancer cells. To better understand the role of DNMT3B binding, DNA methylation, and histone modifications at enhancer regions in stilbenoids-mediated downregulation of gene transcription, we have performed chromatin immunoprecipitation (ChIP) followed by next-generation sequencing to analyze binding events of DNMT3B and occupancy of H3K36me3 at a genome-wide scale. We then elucidated whether altered DNMT3B and H3K36me3 enrichment are linked to changes in transcription factor binding and gene transcriptional activity.
2. Overview of DNMT3B Binding in Highly Invasive MCF10CA1a Breast Cancer Cells in Response to Pterostilbene (PTS)
To understand a possible functional role of DNMT3B in mediating epigenetic effects of stilbenoid polyphenols, we performed ChIP for DNMT3B in invasive MCF10CA1a breast cancer cells upon 9-day treatment with PTS at 7 μM concentration, followed by next-generation sequencing. Upon analysis of DNMT3B ChIP-seq data, we identified statistically significant changes in DNMT3B binding in 3314 peaks throughout the genome. Of those peaks, 1939 peaks were enriched with DNMT3B upon PTS (Figure 1A).
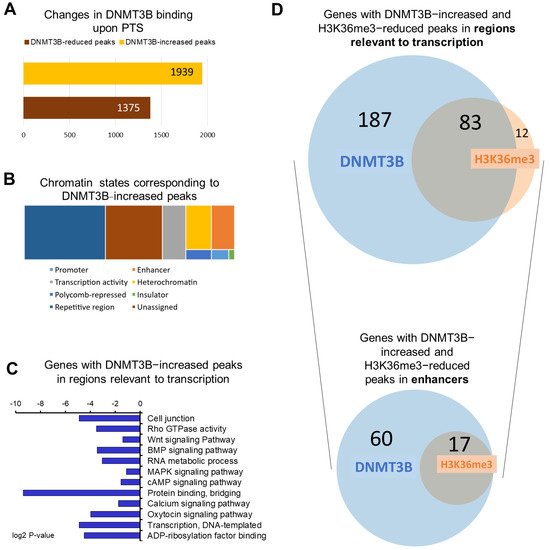
Figure 1. Overview of genome−wide changes of DNMT3B binding in response to PTS. (A) A bar chart depicting the number of changes in DNMT3B binding in MCF10CA1a breast cancer cells treated with 7 μM PTS for 9 days. DNMT3B binding was assessed by ChIP sequencing. (B) Chromatin states associated with DNMT3B-increased peaks were determined using Broad ChromHMM data from human mammary epithelial cells (HMEC) available on USCS Genome Browser (hg19). Peaks could correspond to active, weak, or poised promoters, strong or weak enhancers, putative insulators, active or weak transcription, Polycomb−repressed regions, heterochromatin, or repetitive regions. (C) The 270 genes with DNMT3B-increased peaks in regions relevant to transcription upon PTS (i.e., promoters, enhancers, insulators, Polycomb-repressed regions, or heterochromatin) were subjected to Gene Ontology (GO) function and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis using DAVID Knowledgebase. (D) Venn diagrams present an overlap between genes with DNMT3B−increased and H3K36me3-reduced peaks that were located in regions relevant to transcription and specifically in enhancer regions. The proportionally scaled Venn diagrams were generated using tools on www.stefanjol.nl/venny (accessed on 28 May 2021).
Using the Broad ChromHMM track associated with human mammary epithelial cells (HMEC) available on the USCS Genome Browser, DNMT3B-increased peaks were annotated to corresponding chromatin states. The peaks were mostly located in repetitive elements (38.5% of peaks), which is in accordance with the well-established DNMT3B function in transcriptional repression of these elements, crucial for genomic stability [29][39]. While 27% of peaks were located in regions with unspecified chromatin state, the remaining 665 peaks were found in regions important for regulation of gene transcription, including promoters, enhancers, insulators, Polycomb-repressed regions, or heterochromatin. (Figure 1B).
Using gene ontology (GO) and KEGG tools in the DAVID knowledgebase database, we performed functional and signaling pathway analyses of genes that contained at least one of the 665 DNMT3B-enriched peaks within regions relevant to transcription (Figure 1C). We identified 270 unique genes in this group. We found that these genes are implicated in signaling pathways commonly upregulated in cancer (Wnt, MAPK, BMP, mTOR, NOTCH, PI3K/Akt), in DNA replication, recombination and repair, cell junction, actin cytoskeleton, regulation of transcription, and calcium ion transmembrane transport (Figure 1C). Thereafter we refer to the 270 genes as DNMT3B-enriched target genes.
A thorough analysis of the DNMT3B-enriched target genes revealed candidates with oncogenic functions, including NOTCH2NL, PANX1, PVT1, and JAK2. Of note, NOTCH2NL is an oncogene activating Notch signaling by direct interaction with NOTCH2, thereby promoting proliferation and self-renewal [30][40]. PANX1 overexpression has been associated with a worse prognosis in breast and liver cancer patients, which mechanistically is linked to PANX1-dependent enhancement of epithelial–mesenchymal transition and thus cell invasion [31][32][41,42]. PVT1 is a long noncoding RNA that is commonly overexpressed in breast cancer and has been implicated in the regulation of MYC oncogene [33][34][43,44], while JAK2 is a tyrosine kinase activating cancer-driving JAK/STAT signaling pathway [35][45].
Among 665 peaks assigned to chromatin states that are relevant to gene transcription, 201 peaks were located within promoters or enhancers, both of which are the core cis-regulatory elements. Interestingly, the majority of those 201 peaks are in enhancer regions (i.e., 173 peaks corresponding to 77 unique genes). The 77 genes with enhancers that gained DNMT3B binding upon PTS fell into several functional categories: (1) oncogenes such as PITPNC1 [36][46], NOTCH2NL [30][40], TNNT2 [37][47], and ZP4 [38][48], (2) long noncoding RNAs (lncRNAs) such as DANT2, LINC00910, and LOC102724511, (3) microRNAs miR4477A and miR4477B, (4) small noncoding RNAs RNVU1-18 and SNAR-A14), (5) pseudogenes LOC100130331, LOC102724580, and (6) epigenetic regulator SMARCA4, which is the ATPase of the chromatin-remodeling SWI/SNF complexes [39][49]. Many of those genes are linked to functions and processes that drive cancer. For instance, serine/threonine protein kinase NLK was demonstrated to localize in the nucleus of breast cancer cells and protect the cells from apoptosis [40][50]. The tumorigenic function of the SREBF1 transcription factor, which was found to be overexpressed and positively associated with metastasis and poor prognosis in breast cancer patients, has been linked to oncogenic activation of the PI3K/AKT/mTOR signaling pathway [41][51]. PITPNC1 has been shown to be overexpressed in metastatic breast, colon, and melanoma cancers [36][46], and SMARCA4 is a marker of poor prognosis in many types of cancer [39][49]. We also identified a group of noncoding RNAs with DNMT3B-enriched enhancers (e.g., LINC00910). Although understanding of functions of noncoding RNAs in carcinogenesis is limited, there is evidence indicating that noncoding RNAs impact a wide range of biological processes such as metabolism [42][52], immune response [43][53], and development [44][54], all of which have been found to be dysregulated during carcinogenesis.
3. H3K36me3 Occupancy Is Reduced at Regions Enriched with DNMT3B in Response to PTS
In order to test whether DNMT3B binding is linked to changes in the activity of enhancers, we performed ChIP-seq with antibodies recognizing H3K27ac, a histone mark at active enhancers, in our preliminary experiment. However, we did not detect any apparent changes in H3K27ac upon PTS. Therefore, we proceeded with assessing PTS impact on the distribution of H3K36me3, which is a unique mark that can distinguish active enhancers from intermediate and poised enhancers and is associated with transcriptional activity [13]. Using ChIP-seq, we found 673 peaks, which showed statistically significant differential enrichment with H3K36me3 in response to PTS. Among those peaks, there were 324 peaks with a reduced enrichment in cells treated with PTS, as compared with vehicle-treated cells. The analysis of the chromatin states showed that 135/324 peaks are located in regions relevant to transcription and correspond to 95 unique genes. Our hypothesis, formulated based on our previous study [20], was that DNMT3B binds enhancers of oncogenes, catalyzes DNA methylation, and reduces the activity of enhancers upon PTS, which consequently downregulates gene expression. Thus, we focused on finding an overlap between genes with DNMT3B-increased peaks and genes with H3K36me3-reduced peaks, with the latter mark indicating reduced activity of an enhancer in a given gene. As shown in Figure 1D, we identified 83 genes that had reduced H3K36me3 mark and increased DNMT3B binding within regions relevant to transcription. Importantly, among genes with H3K36me3-reduced peaks, 17 genes were associated with changes in enhancers, and all 17 genes were also found within genes containing DNMT3B-increased peaks (Figure 1D and Figure 2A). For all of the 17 genes, reduced H3K36me3 occupancy and increased DNMT3B binding were detected within 200 bp in the same enhancer region (Figure 2B). We next focused on the 17 genes for further analysis and refer to these genes as ‘candidate genes with epigenetically targeted enhancers in response to PTS’ (Figure 2B). Among them, there were known oncogenes, PITPNC1 [36][46], TNNT2 [37][47], and ZP4 [38][48], and lncRNAs with a putative oncogenic role, such as DANT2 and LINC00910, that we mentioned in the previous paragraph. We also found several genes that may function as oncogenes within the context of breast cancer. For example, CUX1 encodes for a transcription factor that has been demonstrated to bind to the Snail promoter, which ultimately leads to increased epithelial-to-mesenchymal transition (EMT), tumor migration, and invasion [45][55]. Recently, active CUX1 has been shown to be upregulated in triple-negative breast cancer, and upon knockdown of CUX1, there was increased estrogen receptor ERα and drug sensitivity [46][56]. Another candidate, RYR2, encodes for a ryanodine receptor, a component of a calcium channel, and it has been previously demonstrated that altered cellular calcium homeostasis contributes to the EMT in breast cancer [47][48][57,58]. Indeed, it was described that upon the EMT of MDA-MB-468 breast cancer cells, there was a drastic increase in RYR2, indicating that this receptor may play an important role in the process.
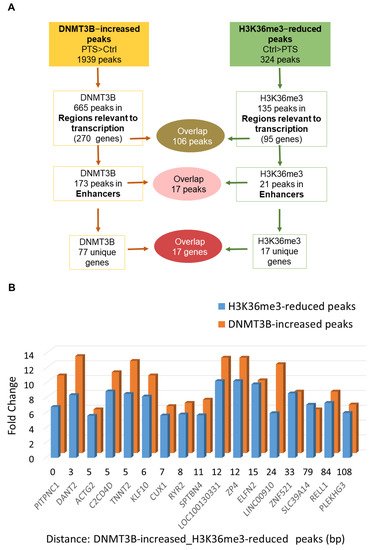
Figure 2. Characterization of regions and genes that contain overlapping DNMT3B-increased and H3K36me3-reduced peaks in response to PTS. (A) Schematic of the analysis of peaks and genes associated with DNMT3B-increased and H3K36me3-reduced peaks in MCF10CA1a breast cancer cells treated with 7 μM PTS for 9 days. The overlap between peaks located within regions relevant to transcription (i.e., promoters, enhancers, insulators, Polycomb-repressed regions, or heterochromatin) and specifically enhancers is depicted along with the number of corresponding genes. (B) The 17 overlapping peaks located in enhancer regions and the corresponding genes are shown in the bar chart that computes a difference (fold change) in DNMT3B binding and H3K36me3 enrichment between PTS-treated and control cells (vehicle-treated, ethanol), along with the distance between DNMT3B-increased and H3K36me3-reduced peaks in a given gene. The presented results are based on ChIP-seq analyses.
4. Discussion
A crucial role of enhancers in gene transcriptional activity has long been an area of interest, with a more recent focus on the contribution of epigenetic components, including DNA methylation, in regulating the activity of enhancer regions [10][14][15][10,14,15]. Dysregulation of DNA methylation patterns and aberrant expression of DNMTs have been observed across multiple cancer types [49][50][59,60]. For instance, tumor suppressor genes are often methylated and silenced during carcinogenesis, whereas oncogenes lose methylation within their regulatory regions, including enhancers, and become actively transcribed [5][20][5,20]. Our group has shown that stilbenoids mediate transcriptional repression of MAML2 through altering epigenetic patterns at the enhancer region of this oncogene [20]. DNMT3B was suggested to be a key player in MAML2 hypermethylation and silencing in response to stilbenoids.
In the present study, we therefore investigated epigenetic marks and the interaction of DNMT3B with DNA at enhancer regions in response to PTS in breast cancer cells. Following ChIP-seq, we found 173 DNMT3B-increased peaks within enhancer regions corresponding to 77 unique genes (Figure 1D and Figure 2A). Functional analyses show enrichment of these genes with oncogenic processes and pathways, for example, PITPNC1 is involved in metastasis [36][46], NOTCH2NL is from the NOTCH signaling pathway [30][40], and SREBF1 is a transcription factor linked to oncogenic PI3K/AKT/mTOR pathway [41][51]. Of importance, although DNMT3B’s role in the regulation of gene transcription appears to be context-dependent, the enzyme has previously been shown to target genes with oncogenic functions for methylation and silencing in different cancers, including breast cancer [51][52][53][54][61,62,63,64]. Interestingly, in our ChIP-seq data, reduced binding of DNMT3B in response to PTS seems to occur at different genes than the increased binding. Among genes associated with DNMT3B-reduced peaks within gene regulatory regions, we found strong candidates associated with inhibition of cancer proliferation, migration, and metastasis, and activation of apoptosis. For instance, we identified CHRDL1, the downregulation of which was associated with poor survival and mechanistically with cancer progression and metastasis, indicating the tumor-suppressing role of CHRDL1 [55][65]. Other interesting candidates were NOX5, whose activation was demonstrated to inhibit cancer stem cell formation through ROS generation [56][66], and SALL3, which was reported to directly inhibit DNMT3A activity and consequently cause DNA hypomethylation and activation of tumor suppressor genes [57][67]. Thus, our findings imply that DNMT3B may be implicated in the methylation and silencing of oncogenes in normal cells.
Interestingly, previous studies report delocalization of DNMT3B in cancer and link this phenomenon, at least partially, to oxidative stress and DNA repair mechanisms [58][59][68,69]. It is suggested that oxidative stress leads to DNA breaks and activation of the DNA repair pathway, which consequently recruits DNMT3B to sites of DNA damage [58][59][68,69]. When the repair system is overloaded, it may cause a permanent shift in DNMT3B localization, resulting in a transition from the silencing of oncogenes to the methylation and silencing of tumor suppressor genes. As PTS is known to regulate the antioxidant defense network [60][61][62][70,71,72], we could hypothesize that PTS, by decreasing oxidative stress, may alter DNMT3B localization and shift it back to regions where DNMT3B typically binds in normal cells [22]. This interesting concept will be further explored in our future studies.
We found that 17 of the enhancer regions with DNMT3B-increased binding in response to PTS overlap within 200 bp with reduced occupancy of H3K36me3 (Figure 2). H3K36me3 is a very important histone mark linked to transcriptional activation and elongation and distinguishing active from intermediate or poised enhancers [13]. H3K36me3 was found to be abundant along with phosphorylated RNA polymerase II at actively transcribed genes, and, as such, it is an excellent mark of transcribed genes [13]. Nearly 31% of genes with DNMT3B-increased peaks in regions relevant to transcription lost H3K36me3 in response to PTS (Figure 1D). This clearly indicates a decrease in the transcriptional activity of those genes upon PTS.
Genes assigned to the 17 enhancers included oncogenes and pro-metastatic genes (PITPNC1 [36][46], TNNT2 [37][47], ZP4 [38][48], CUX1 [45][55], RYR2 [47][48][57,58]), and putative oncogenes (DANT2 and LINC00910). Indeed, enrichment of ectopic H3K36me3 at oncogenes has previously been reported in different cancers, including breast cancer, and associated with gene upregulation [63][64][65][66][67][73,74,75,76,77]. Redistribution of H3K36me3 across intergenic regions and low-expressing genes was associated with aberrantly upregulated expression [68][78]. As H3K36me3 is known to antagonize Polycomb repressive complex 2 (PRC2)-mediated H3K27 methylation [69][70][79,80], perhaps loss of H3K36me3 at enhancers of oncogenes in response to PTS results in increased PRC2-mediated gene repression. Another interesting aspect is that genes expressed in a cell type-dependent manner are known to be targeted by de novo DNMT activity and methylated as a consequence [71][81]. DNA methylation eventually replaces dynamic histone marks to provide stable silencing of these genes upon differentiation [71][81]. Indeed, along with increased occupancy of DNMT3B, we observed DNA methylation and decreased expression of PITPNC1 and LINC00910 (Figure 3).
In addition, almost half of the overlapping DNMT3B-increased and H3K36me3-reduced peaks located in regions relevant to transcription (i.e., promoters, enhancers, insulators, Polycomb-repressed regions, or heterochromatin) also lost OCT1 binding in response to PTS (Figure 4A, Supplementary Table S2 with enhancers). This finding raised the possibility that OCT1 plays a role in the regulation of transcription of corresponding genes as all the changes refer to reduced transcription. Indeed, OCT1 depletion in MCF10CA1a breast cancer cells led to a profound downregulation of PITPNC1 and LINC00910 (Figure 4B) and exerted anti-cancer effects [20]. Taking into account that the overlapping DNMT3B-increased and H3K36me3-reduced peaks within regions relevant to transcription are assigned to genes enriched with cancer-driving functions and pathways (Figure 1C), our results are in agreement with existing literature. The pro-tumorigenic function of OCT1 has been demonstrated in different cancer types, and OCT1 has been shown to regulate genes associated with cell metabolic function, proliferation, oxidative stress, and immune modulation, all of which are interconnected with a process of tumorigenesis [72][73][82,83]. DNMT3B- and OCT1-mediated mechanisms, through which stilbenoids modify DNA methylation and impact the expression of genes involved in cancer, are an emerging research area.
The overlap between OCT1-reduced peaks and DNMT3B-increased and H3K36me3-reduced peaks also suggests that OCT1 may matter for specificity in DNMT3B binding to those regions. Indeed, several pieces of evidence indicate that DNMT3B recruitment may be directed by the recognition of transcription factors [29][74][39,84]. It was reported that certain transcription factors, for example, E2F6, NR6A1, and PU.1, act as positive regulators of DNMT3B recruitment, leading to silencing of their target genes [74][84], whereas CTCF and SP1 were shown to block de novo DNA methylation at the target regions [74][84].
In terms of DNMT3B recruitment to the target regions, another interesting hypothesis involves histone modifications, such as H3K36me3, as was suggested by other groups [75][76][85,86]. DNMT3B selectively bound the bodies of transcribed genes and led to their preferential methylation, which was mediated through recognition of H3K36me3 in mouse embryonic stem cells [75][85]. Recruitment of DNMT3B to cell-type-specific actively transcribed enhancers, followed by their hypermethylation, was also demonstrated to be mediated by recognition of H3K36me3 in human epidermal stem cells [76][86]. Depletion of SETD2, an enzyme responsible for H3K36me3, particularly affected DNA methylation at H3K36me3 sites, implying that H3K36me3 is key to guide loci-specific recruitment of DNMT3B [75][85]. On the other hand, as we mentioned before, loss of H3K36me3 may result in increased PRC2-mediated gene repression [69][70][79,80]. Therefore, the increase in DNMT3B binding and DNA methylation within regions of decreased H3K36me3 upon PTS may just be a consequence of decreased expression, irrespective of H3K36me3 recruitment activity of DNMT3B. Those interesting interactions between epigenetic enzymes and proteins and the consequences of such interactions for the regulation of gene expression should be further explored in vivo. Of importance, current technological advances in cell sorting allow for the isolation of single cells from tumors [77][78][79][87,88,89]. It makes the in vivo approach highly feasible. It would also be critical to elucidate whether DNMT3B plays similar roles in other cancers, especially other aggressive cancers characterized by activation of oncogenes (e.g., pancreatic cancer [80][90]), and whether PTS effects on DNMT3B binding and histone mark occupancy occur in different cancer types.