Circular RNAs (circRNAs) are a new class of endogenous non-coding RNAs with covalent closed loop structure. Researchers have revealed that circRNAs play an important role in human diseases. As experimental identification of interactions between circRNA and disease is time-consuming and expensive, effective computational methods are an urgent need for predicting potential circRNA–disease associations. In this study, we proposed a novel computational method named GATNNCDA, which combines Graph Attention Network (GAT) and multi-layer neural network (NN) to infer disease-related circRNAs. Specially, GATNNCDA first integrates disease semantic similarity, circRNA functional similarity and the respective Gaussian Interaction Profile (GIP) kernel similarities. The integrated similarities are used as initial node features, and then GAT is applied for further feature extraction in the heterogeneous circRNA–disease graph. Finally, the NN-based classifier is introduced for prediction. The results of fivefold cross validation demonstrated that GATNNCDA achieved an average AUC of 0.9613 and AUPR of 0.9433 on the CircR2Disease dataset, and outperformed other state-of-the-art methods.
- circRNA–disease associations
- graph attention network
- multi-layer neural network
1. Introduction
cap and a 3
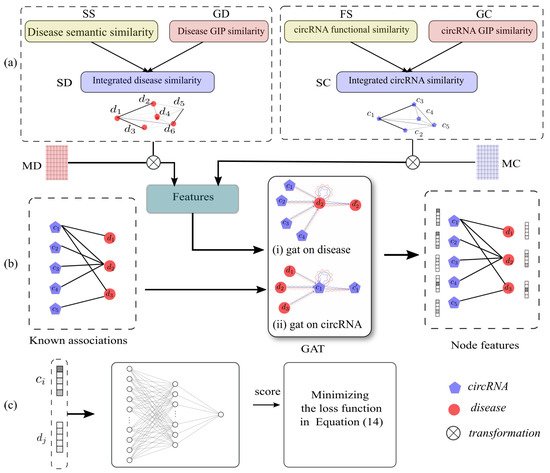
-
We proposed an end-to-end framework for inferring disease-related circRNAs, which can effectively and accurately infer the potential associations between circRNAs and diseases.
-
We made use of GAT to extract low-dimensional dense representations of circRNAs and diseases, and these presentations had rich structural and semantic information of the heterogeneous circRNA–disease graph.
-
We proposed a NN-based classifier, and applied a sampling strategy to construct balanced samples. In addition, we designed cross-entropy loss with L2 regularization to make the training process fast and robust.
-
We demonstrated the predictive performance of our method by extensive experiments via fivefold cross validation and case studies, and achieved competitive results on CircR2Disease and circRNADisease datasets.
2. Case Studies
To further evaluate the prediction ability of our proposed method, we performed two case studies in this section. We trained GATNNCDA on CircR2Disease dataset [22], and then verified the candidates on circRNADisease [19] and circAtlas v2.0 [24] datasets. The first case study was conducted on breast cancer, which is one of the most common cancers in women. In particular, we constructed the positive samples with all known associations between circRNAs and diseases in the CircR2Disease. Meanwhile, we randomly chose the same number of negative samples from the unknown associations. Based on these training samples, we built the GATNNCDA and calculated the scores between breast cancer and each circRNA. Finally, we selected the top 20 related circRNAs for analysis. As shown in Table 4, 18 of the top 20 are confirmed by the validation datasets. The other two candidates have been verified in the recently published literature.Table 14. Top 20 predicted circRNAs related to Breast cancer based on circR2Disease dataset.Rank circRNA Evidence Rank circRNA Evidence 1 hsa_circ_0007534 II 11 hsa_circ_0068033 I ; II 2 hsa_circ_0011946 II 12 circamotl1hsa_circ_0004214 I ; II 3 hsa_circ_0093859 II 13 hsa_circ_0006528 I ; II 4 circrna-000911 II 14 hsa_circ_0002874 I ; II > 5 circrna-001283 PMID:29431182 15 hsa_circ_0001667 I ; II 6 circrna-001175 II 16 hsa_circ_0085495 I ; II 7 circrna-100438 PMID:29431182 17 hsa_circ_0086241 I ; II 8 hsa_circ_0001982 I ; II 18 hsa_circ_0092276 I ; II 9 hsa_circ_0001785 I 19 hsa_circ_0003838 I ; II 10 hsa_circ_0108942 I ; II 20 circvrk1 I ; II I, II denote circRNADisease, circAtlas v2.0.The second case study is performed on hepatocellular carcinoma. It is the most common form of liver cancer, with a higher incidence in patients with long-term liver diseases [33][44]. We utilized GATNNCDA to calculate the correlation score with circRNAs and then sorted by descending order. The top 20 hepatocellular carcinoma related cirRNAs are listed in Table 25. We can see that 10 of the top 20 are verified by the validation datasets, and the other eight candidates have been conformed in relevant literature, e.g., hsa_circ_0000520 is one of the three circRNAs that showed significantly different expression levels in HCC tissues [14]. Therefore, the unknown associations with high scores are likely to be correlated.I, II denote circRNADisease, circAtlas v2.0.Table 25. Top 20 predicted circRNAs related to hepatocellular carcinoma based on circR2Disease dataset.Rank circRNA Evidence 1 circc3p1 II 2 hsa_circ_0067531 II 3 circarsp91hsa_circ_0085154 II 4 circmto1hsa_circrna_0007874hsa_circrna_104135 II 5 hsa_circ_0005986 I ; II 6 hsa_circrna_100338circsnx27 PMID:28710406 7 hsa_circrna_104075 I ; II 8 hsa_circrna_102049 PMID:28710406 9 circrna_000839 II 10 circzkscan1hsa_circ_0001727 I ; II 11 hsa_circ_0004018 I ; II 12 hsa_circ_0005075 II 13 hsa_circrna_100571 PMID: 29609527 14 hsa_circrna_400031 PMID:29609527 15 hsa_circrna_102032 PMID: 29609527 16 hsa_circrna_103096 PMID:29609527 17 hsa_circrna_102347 PMID:29609527 18 hsa_circrna_000167hsa_circ_0000518 unknown 19 hsa_circ_0000520 PMID:27258521 20 hsa_circ_0000172 unknown 3. Conclusions
Cumulative evidence has shown that circRNAs play an important role in progression of human diseases, and are suitable as promising disease biomarkers for prevention, diagnosis and treatment. As traditional biological identification is very costly and time-consuming, more and more computational methods have been introduced in this field. In this study, we proposed a novel computational method called GATNNCDA for predicting potential circRNA–disease associations. GATNNCDA achieved a better performance than other state-of-the-art methods by combining similarity integration, graph attention network and multi-layer neural network. In particular, we performed fivefold CV for evaluation, and obtained the best performance of AUC of 0.9742, AUPR of 0.9707. The average values of AUC and AUPR for under 50 experiments were 0.9613 and 0.9452. Furthermore, case studies on breast cancer and hepatocellular carcinoma have also demonstrated that GATNNCDA can be a useful tool for predicting potential disease-related circRNAs.However, GATNNCDA still has some limitations. The initial node features may not be perfect. Recall that similarity integration as initial node representations would affect the final performance. Nonetheless, known interactions between circRNA–disease associations are insufficient. In addition, circRNA functional similarity and GIP similarity may be inaccurate. Therefore, more biological information such as circRNA–miRNA association or circRNA sequence will be used for further study to construct more accurate node features, especially for some unseen circRNAs. Furthermore, the NN-based classifier of GATNNCDA requires negative samples for training, which are rarely reported in the literature. Randomly sampling from the unknown associations in a CircR2Disease dataset would introduce bias. In the future, we will seek a better negative sampling strategy to promote the performance of GATNNCDA.