Telomerase reverse transcriptase (TERT) has been established to possess diagnostic value in leukemia as most adult cells do not express high levels of telomerase. Indeed, studies have shown that prognosis is not favorable in patients who have leukemias expressing high levels of telomerase. Recent research has indicated that targeting of this gene is able to control the survival of malignant cells and therefore offers a potential treatment for TERT-dependent leukemias.
- hTERT
- leukemia
- gene regulation
- cancer
- hematological malignancy
1. Introduction
The human telomerase reverse transcriptase gene (hTERT) spans over a 40 kb DNA region and consists of 16 exons and 15 introns [1]. This gene produces a polypeptide with a length of 1132 amino acids which is then transformed to a 130kD functional TERT protein [2]. The four critical functional domains in TERT comprise the N-terminal regulatory domain, the RNA binding domain, the reverse transcriptase domain, and the C-terminal dimerization domain [3]. Owing to its complexity, TERT is regulated at various levels which include transcriptional, post-transcriptional, and post-translational mechanisms [4,5,6][4][5][6].
The primary function of this protein is in the maintenance of telomere length. Telomeres cap the ends of chromosomes with repetitive 5′-TTAGGG-3′ conserved sequences to maintain the integrity of genetic information [7]. With each successive round of cell proliferation, 50 to 200 base pairs of DNA material are dissociated from the ends of the chromosomes due to the loss of RNA primers situated on the lagging strand of Okazaki fragments. This phenomenon leads to the progressive shortening of telomeres which limits the potential of somatic cells to divide [8,9][8][9]. Certain cell types, such as stem cells and cancer cells, are able to express telomerase to replenish telomeric ends, thereby permitting continuous self-renewal [10,11,12][10][11][12]. Telomerase exerts its DNA polymerizing function through the formation of a complex consisting of two main subunits: the catalytic protein hTERT and the human telomerase RNA component (hTERC) subunit [13].
2. Transcriptional Regulation of TERT
The promoter of TERT is a very dynamic region. It is GC rich and lacks the TATA or CCAAT recognition sequence for the transcription start site (TSS) location by RNA polymerase 11. However, this is compensated for by an initiator like CCTCTCC sequence. It also contains two E boxes for Myc binding, several GC boxes for Sp1 binding, and a CCAC box [6]. These motifs make the promoter region an excellent hub for the binding of various repressors and activators [5,6][5][6].
2.1. Negative Transcriptional Regulators of TERT
Negative transcriptional regulators of TERT can either bind directly to the promoter or interact by forming complexes to downregulate TERT expression. Direct acting repressors include retinoblastoma (RB), myeloid-specific zinc finger protein (MZF2), activator protein 1 (AP1), Wilms tumor 1 (WT1), and menin [23,24,25][14][15][16]. Transcriptional repressors exert their effect on TERT expression by forming complexes and interacting with other proteins including Mad1, the HTLV-1 oncogene TAX, tumor suppressors BRCA1/Nmi and p53, interferon regulatory factor 1 (IRF1), and transforming growth factor β (TGFβ) [26,27,28,29,30][17][18][19][20][21]. Mad1 has been identified to form a complex with Max (Mad1/Max) and binds to the E box region located at the promoter to prevent TERT expression [26,31,32][17][22][23]. Similarly, it was found that TAX also represses TERT expression by E-box binding [27][18]. Receptor Ck, on the other hand, has been determined to regulate TERT via interactions with protein kinase C [33][24].
2.2. Positive Transcriptional Regulators of TERT
2.3. Conditional Transcriptional Regulators of TERT
There are some regulators, such as transcription factors E2F and specificity protein 1 (Sp1), upstream stimulatory factors (USF1/2), and tumor suppressor p73, which act as both repressors and activators of TERT expression depending on the condition. E2F acts as a TERT repressor in cancer cells; however, the condition is reversed in normal cells where it activates the expression of TERT. This transcription factor controls TERT regulation by binding to the GC box region on the promoter [41,42][28][29]. USF1/2 on the other hand acts as a repressor in normal cells and activator in cancer cells. This is achieved by E box binding on the promoter [43,44][30][31].3. Post Transcriptional Regulation of TERT
TERT Regulation by microRNAs
4. Post Translational Regulation of TERT
Post-translational modifications of hTERT could modify protein stability, subcellular localization, and ultimately, enzyme activity [53][36]. To date, studies have identified that post-translational regulation of hTERT mainly involve phosphorylation and ubiquitination by kinases and ligases respectively [4,53,66][4][36][40]. Both phosphorylation and ubiquitination give rise to prominent effects by positively and negatively regulating hTERT activity [67][41].
In phosphorylation, kinases such as protein phosphatase 2A (PP2A) interact with c-Abl tyrosine kinase protein, leading to a threefold reduction of telomerase activity by dephosphorylation [53,68,69][36][42][43]. On the contrary, protein kinase B (an AKT kinase) increases telomerase activity through TERT phosphorylation-dependent activation of the PI3K/Akt/mTOR pathway [68,69][42][43]. Similar to protein kinase C, they phosphorylate hTERT by PKC isoenzymes α, β, δ, ε, and ζ to enhance telomerase activity, which is increased along with nuclear accumulation of hTERT [61,66,69,70,71][33][40][43][44][45]. In addition, kinases including kinase interacting protein (KIP), c-jun, and mitogen activated protein kinase (MAPK) also regulate post-translational modifications of hTERT. KIP regulation involves interactions with the upstream kinase domain of DNA–PKcs to improve telomerase activity in human cells, while, MAPK upregulate hTERT in a serum and pH-dependent manner within the hypoxic environment of solid tumors [5,72][5][46].
5. TERT Dysregulation in Leukemias
Studies on the various forms of leukemia have disclosed the dysregulation of TERT gravely affects the prognosis of the disease and is known to exert its mechanism of action via a plethora of modifications including epigenetics, mutations, amplifications, structural variants, and influences on oncogenes. The prevalence of TERT dysregulation remains to be determined in each subtype of leukemia and is also highly dependent on the population and recruitment criteria of each study. An early study has indicated that it affects approximately half of AML (53.3%) patients [80][47], while another report indicated that TERT dysregulation is present in all acute promyelocytic leukemia (APL, AML-M3) patients in their cohort [81][48]. Other studies have shown that hTERT dysregulation was only observed in limited cases of relapsed childhood ALL [82][49], while a study in Saudi Arabia disclosed that hTERT mutations are not linked to predisposition to childhood acute leukemia [83][50]. In terms of relevance in disease pathogenesis, studies have shown that in adult T-cell leukemia/lymphoma (ATLL), the activation of telomerase is required for the disease development and progression, while in AML and CML it is only required for maintenance and not initiation [15,84,85][51][52][53].
5.1. Chronic Myeloid Leukemia (CML)
Mechanism of TERT dysregulation was reported to occur via TERT promoter methylation. However, contrary to canonical research, DNA methylation would increase TERT activity, as methylation occurs on the genomic region bordering the TERT promoter. This results in the inaccessibility of repressor on this site, which is proximal to the TERT promoter, thereby increasing the affinity of TERT promoter binding of crucial activators leading to TERT hyper-expression (Figure 1A).
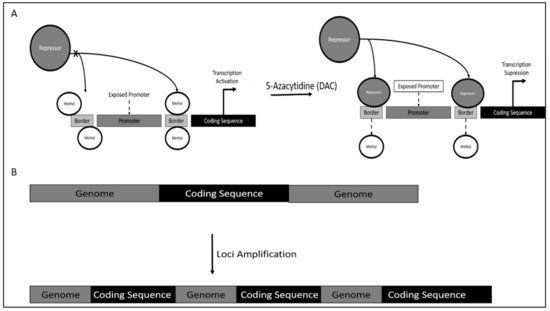
5.2. Chronic Lymphoid Leukemia (CLL)
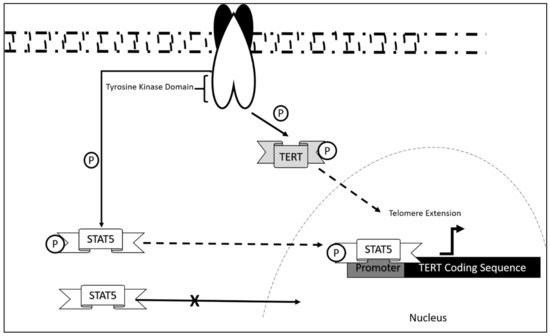
The mechanism of dysregulation of TERT in CLL is still largely unexplored. However, correlation studies report that telomeric length serves as an initial prognostic marker in CLL disease progression [95][56]. This was also associated with telomerase/telomere associated proteins expression, which also contribute as markers in the diagnosis of CLL onset. Research showed that significant decrease in telomeric length was observed in chromosomes 13p, 12, 11p, and 17p in which hypomethylation was also present. These aberrations were significant in CLL patients as compared to normal controls. Upregulation of telomerase/telomere-associated proteins was also observed in patients at early disease onset [96][57]. As in the case of most leukemia, TERT activation occurs via STAT5 mediated binding and its phosphorylation state [89,97,98][58][59][60]. This mechanism, although not studied extensively in CLL, may be a contributing mechanism as to how CLL progress in addition to the upregulated telomerase/telomere-associated proteins and telomeric length dysregulation.
Furthermore, in terms of DNA methylation status it was found that methylation of TERT promoter was crucial in different stages of leukemia development as it confers different functional advantages.
Dysregulation of TERT in CLL was also observed via single nucleotide polymorphisms (SNPs). Different SNPS led to different potential in TERT induced self-renewal [102][61].
Additionally, variation within the TERT and TERT C gene also contributed to an increase in telomeric ends. It was found that TERTC rs10936599 SNP with C allele had a significant association with CLL development as high allele frequency was observed in CLL patients.
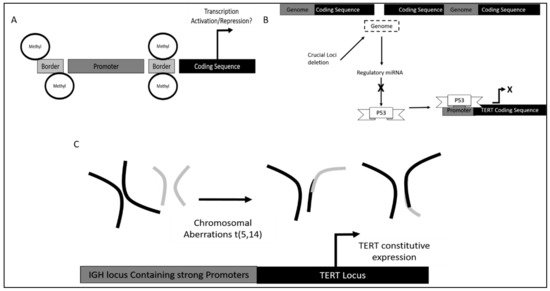
5.3. TERT Dysregulation and Clinical Implications in Acute Leukemias
Acute leukemia refers to an aggressive form of leukemia that typically progresses rapidly over a short period of time. It can be further subdivided into acute myeloid (AML) and acute lymphoid leukemia (ALL) [113][66]. TERT dysregulation that has been identified as one of the contributors to the pathogenesis of leukemia and has been reported in both AML and ALL.
5.3.1. TERT Dysregulation and Clinical Implications in Acute Myeloid Leukemia
Epigenetic regulation of TERT has been the focus of many studies as it is believed to aid in the malignant transformation of cells [54][32]. Hypermethylation of the TERT promoter results in TERT transcription activation that subsequently leads to higher telomerase activity [124][67]. According to a study by Zhao et al. (2016), TERT proximal promoter and a partial exon 1 (TERTpro/Ex1) region exhibits CpG site hypermethylation in AML cell lines and primary blasts [125][68]. This distinct methylation profile could serve as a prognostic factor in AML as it is associated with shorter overall survival and drug resistance. Apart from the above regulation, TERT is also regulated by oncogenic signaling FLT3-ITD (FMS-Like Tyrosine Kinase 3 Internal Tandem Duplication). FLT3, a type of receptor tyrosine kinase is often mutated in AML with internal tandem duplications within the juxtamembrane domain. Compelling evidence has implicated the role of FLT3-ITD mutation in leukemogenesis especially in the case of AML [126][69]. Recently, it has come to light that FLT3-ITD mutation could induce the expression of TERT, resulting in increased telomerase activity that contributes to AML pathogenesis [127][70]. Another method of telomeric elongation identified in promyelocytic leukemia (PML) (a form of AML) is alternative lengthening of telomeres (ALT) [128][71].
Conventional chemotherapy is one of the therapeutic approaches used to treat AML patients, whereby it is common that a combination of chemotherapeutic drugs is used. Each of the chemotherapeutic drug aims to target and inhibit the cancer cells via various mechanisms. One of the mechanisms targeted by a commonly used drug for AML, 5-azacytidine (5-AZA) is DNA methylation. 5-AZA, a DNA methyltransferase inhibitor (DNMTI), has been shown to downregulate TERT expression and decrease telomerase activity in AML patient samples and cell lines [131][72].
TERT-targeted immunotherapy has also become the subject of many cancer therapeutic studies. A study by Sandri et al. (2017) demonstrated that TERT865-873-specific, TCR-engineered T-cell lymphocytes were able to halt AML progression in vivo [134][73]. The main idea behind this therapy, known as adoptive T-cell therapy (ACT), is to increase the affinity and specificity of T-cell receptor (TCR) of T cells to tumor associated antigen (TAA). It is rather interesting to note that TERT865-873-specific, TCR-engineered T-cell lymphocytes were able to distinguish between AML blasts and peripheral blood mononuclear cells (PBMC), indicating its high cell recognition specificity.
5.3.2. TERT Dysregulation and Clinical Implications in Acute Lymphoblastic Leukemia
TERT-targeted therapies are quickly gaining popularity as a therapeutic option due to the role of TERT as a universal tumor associated antigen (TAA). BIBR1532, a small biological molecule specific for telomerase inhibition, has been studied by Bashash et al. (2017) for its application in pre-B ALL [141][74]. BIBR1532 was able to suppress TERT expression and telomerase activity in vitro in a dose-dependent manner. In addition, BIBR1532 suppressed the expression of c-Myc that acts as a transcriptional activator of TERT. Doxorubicin, a commonly used chemotherapeutic drug, was found to exert the same inhibitory action as BIBR1532 on TERT and c-Myc. This study also reported that the combination of doxorubicin together with BIBR1532 showed a greater inhibition of TERT and c-Myc transcription as compared to its individual counterparts. Thus, this combination therapy can be an effective therapeutic approach for pre-B ALL.
6. Conclusions and Future Perspectives
References
- Cong, Y.-S.; Wright, W.E.; Shay, J.W. Human Telomerase and Its Regulation. Microbiol. Mol. Biol. Rev. 2002, 66, 407–425.
- Ly, H. Telomere Dynamics in Induced Pluripotent Stem Cells: Potentials for Human Disease Modeling. World J. Stem Cells 2011, 3, 89–95.
- Kelleher, C.; Teixeira, M.T.; Förstemann, K.; Lingner, J. Telomerase: Biochemical Considerations for Enzyme and Substrate. Trends Biochem. Sci. 2002, 27, 572–579.
- Cifuentes-Rojas, C.; Shippen, D.E. Telomerase Regulation. Mutat. Res. 2012, 730, 20–27.
- Depcrynski, A.N.; Sachs, P.C.; Elmore, L.W.; Holt, S.E. Regulation of Telomerase Through Transcriptional and Posttranslational Mechanisms. In Telomeres and Telomerase in Cancer; Cancer Drug Discovery and Development; Hiyama, K., Ed.; Humana Press: Totowa, NJ, USA, 2009; pp. 47–85. ISBN 978-1-60327-879-9.
- Dwyer, J.; Li, H.; Xu, D.; Liu, J.-P. Transcriptional Regulation of Telomerase Activity: Roles of the the Ets Transcription Factor Family. Ann. N. Y. Acad. Sci. 2007, 1114, 36–47.
- Relitti, N.; Saraswati, A.P.; Federico, S.; Khan, T.; Brindisi, M.; Zisterer, D.; Brogi, S.; Gemma, S.; Butini, S.; Campiani, G. Telomerase-Based Cancer Therapeutics: A Review on Their Clinical Trials. Curr. Top. Med. Chem. 2020, 20, 433–457.
- Bernal, A.; Tusell, L. Telomeres: Implications for Cancer Development. Int. J. Mol. Sci. 2018, 19, 294.
- Ramlee, M.K.; Wang, J.; Toh, W.X.; Li, S. Transcription Regulation of the Human Telomerase Reverse Transcriptase (HTERT) Gene. Genes 2016, 7, 50.
- Garbuzov, A.; Pech, M.F.; Hasegawa, K.; Sukhwani, M.; Zhang, R.J.; Orwig, K.E.; Artandi, S.E. Purification of GFRα1+ and GFRα1- Spermatogonial Stem Cells Reveals a Niche-Dependent Mechanism for Fate Determination. Stem Cell Rep. 2018, 10, 553–567.
- Jaiswal, R.K.; Kumar, P.; Kumar, M.; Yadava, P.K. HTERT Promotes Tumor Progression by Enhancing TSPAN13 Expression in Osteosarcoma Cells. Mol. Carcinog. 2018, 57, 1038–1054.
- Lin, S.; Nascimento, E.M.; Gajera, C.R.; Chen, L.; Neuhöfer, P.; Garbuzov, A.; Wang, S.; Artandi, S.E. Distributed Hepatocytes Expressing Telomerase Repopulate the Liver in Homeostasis and Injury. Nature 2018, 556, 244–248.
- Leão, R.; Apolónio, J.D.; Lee, D.; Figueiredo, A.; Tabori, U.; Castelo-Branco, P. Mechanisms of Human Telomerase Reverse Transcriptase (HTERT) Regulation: Clinical Impacts in Cancer. J. Biomed. Sci. 2018, 25, 22.
- Lin, S.Y.; Elledge, S.J. Multiple Tumor Suppressor Pathways Negatively Regulate Telomerase. Cell 2003, 113, 881–889.
- Parodi, S.; Ognibene, M.; Haupt, R.; Pezzolo, A. The Over-Expression of E2F3 Might Serve as Prognostic Marker for Neuroblastoma Patients with Stage 4S Disease. Diagnostics 2020, 10, 315.
- Fujimoto, K.; Kyo, S.; Takakura, M.; Kanaya, T.; Kitagawa, Y.; Itoh, H.; Takahashi, M.; Inoue, M. Identification and Characterization of Negative Regulatory Elements of the Human Telomerase Catalytic Subunit (HTERT) Gene Promoter: Possible Role of MZF-2 in Transcriptional Repression of HTERT. Nucleic Acids Res. 2000, 28, 2557–2562.
- Günes, C.; Lichtsteiner, S.; Vasserot, A.P.; Englert, C. Expression of the HTERT Gene Is Regulated at the Level of Transcriptional Initiation and Repressed by Mad1. Cancer Res. 2000, 60, 2116–2121.
- Gabet, A.-S.; Mortreux, F.; Charneau, P.; Riou, P.; Duc-Dodon, M.; Wu, Y.; Jeang, K.-T.; Wattel, E. Inactivation of HTERT Transcription by Tax. Oncogene 2003, 22, 3734–3741.
- Kanaya, T.; Kyo, S.; Hamada, K.; Takakura, M.; Kitagawa, Y.; Harada, H.; Inoue, M. Adenoviral Expression of P53 Represses Telomerase Activity through Down-Regulation of Human Telomerase Reverse Transcriptase Transcription. Clin. Cancer Res. 2000, 6, 1239–1247.
- Lee, S.-H.; Kim, J.-W.; Oh, S.-H.; Kim, Y.-J.; Rho, S.-B.; Park, K.; Park, K.-L.; Lee, J.-H. IFN-Gamma/IRF-1-Induced P27kip1 down-Regulates Telomerase Activity and Human Telomerase Reverse Transcriptase Expression in Human Cervical Cancer. FEBS Lett. 2005, 579, 1027–1033.
- Lacerte, A.; Korah, J.; Roy, M.; Yang, X.-J.; Lemay, S.; Lebrun, J.-J. Transforming Growth Factor-Beta Inhibits Telomerase through SMAD3 and E2F Transcription Factors. Cell. Signal. 2008, 20, 50–59.
- Oh, S.; Song, Y.H.; Yim, J.; Kim, T.K. Identification of Mad as a Repressor of the Human Telomerase (HTERT) Gene. Oncogene 2000, 19, 1485–1490.
- Xu, D.; Popov, N.; Hou, M.; Wang, Q.; Björkholm, M.; Gruber, A.; Menkel, A.R.; Henriksson, M. Switch from Myc/Max to Mad1/Max Binding and Decrease in Histone Acetylation at the Telomerase Reverse Transcriptase Promoter during Differentiation of HL60 Cells. Proc. Natl. Acad. Sci. USA 2001, 98, 3826–3831.
- Sikand, K.; Kaul, D.; Varma, N. Receptor Ck-Dependent Signaling Regulates HTERT Gene Transcription. BMC Cell Biol. 2006, 7, 2.
- Endoh, T.; Tsuji, N.; Asanuma, K.; Yagihashi, A.; Watanabe, N. Survivin Enhances Telomerase Activity via Up-Regulation of Specificity Protein 1- and c-Myc-Mediated Human Telomerase Reverse Transcriptase Gene Transcription. Exp. Cell Res. 2005, 305, 300–311.
- Katzenellenbogen, R.A.; Egelkrout, E.M.; Vliet-Gregg, P.; Gewin, L.C.; Gafken, P.R.; Galloway, D.A. NFX1-123 and Poly(A) Binding Proteins Synergistically Augment Activation of Telomerase in Human Papillomavirus Type 16 E6-Expressing Cells. J. Virol. 2007, 81, 3786–3796.
- McMurray, H.R.; McCance, D.J. Degradation of P53, Not Telomerase Activation, by E6 Is Required for Bypass of Crisis and Immortalization by Human Papillomavirus Type 16 E6/E7. J. Virol. 2004, 78, 5698–5706.
- Crowe, D.L.; Nguyen, D.C. Rb and E2F-1 Regulate Telomerase Activity in Human Cancer Cells. Biochim. Biophys. Acta 2001, 1518, 1–6.
- Won, J.; Yim, J.; Kim, T.K. Opposing Regulatory Roles of E2F in Human Telomerase Reverse Transcriptase (HTERT) Gene Expression in Human Tumor and Normal Somatic Cells. FASEB J. 2002, 16, 1943–1945.
- Chang, J.T.-C.; Yang, H.-T.; Wang, T.-C.V.; Cheng, A.-J. Upstream Stimulatory Factor (USF) as a Transcriptional Suppressor of Human Telomerase Reverse Transcriptase (HTERT) in Oral Cancer Cells. Mol. Carcinog. 2005, 44, 183–192.
- Goueli, B.S.; Janknecht, R. Regulation of Telomerase Reverse Transcriptase Gene Activity by Upstream Stimulatory Factor. Oncogene 2003, 22, 8042–8047.
- Yuan, X.; Xu, D. Telomerase Reverse Transcriptase (TERT) in Action: Cross-Talking with Epigenetics. Int. J. Mol. Sci. 2019, 20, 3338.
- Farooqi, A.A.; Mansoor, Q.; Alaaeddine, N.; Xu, B. MicroRNA Regulation of Telomerase Reverse Transcriptase (TERT): Micro Machines Pull Strings of Papier-Mâché Puppets. Int. J. Mol. Sci. 2018, 19, 1051.
- Gagnon, J.D.; Kageyama, R.; Shehata, H.M.; Fassett, M.S.; Mar, D.J.; Wigton, E.J.; Johansson, K.; Litterman, A.J.; Odorizzi, P.; Simeonov, D.; et al. MiR-15/16 Restrain Memory T Cell Differentiation, Cell Cycle, and Survival. Cell Rep. 2019, 28, 2169–2181.e4.
- Manasa, V.G.; Kannan, S. Impact of MicroRNA Dynamics on Cancer Hallmarks: An Oral Cancer Scenario. Tumour Biol. 2017, 39, 1010428317695920.
- Jie, M.-M.; Chang, X.; Zeng, S.; Liu, C.; Liao, G.-B.; Wu, Y.-R.; Liu, C.-H.; Hu, C.-J.; Yang, S.-M.; Li, X.-Z. Diverse Regulatory Manners of Human Telomerase Reverse Transcriptase. Cell Commun. Signal. 2019, 17, 63.
- Peng, Y.; Croce, C.M. The Role of MicroRNAs in Human Cancer. Signal Transduct. Target. Ther. 2016, 1, 15004.
- Hou, X.-S.; Han, C.-Q.; Zhang, W. MiR-1182 Inhibited Metastasis and Proliferation of Ovarian Cancer by Targeting HTERT. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 1622–1628.
- Zhao, Q.; Zhai, Y.-X.; Liu, H.-Q.; Shi, Y.-A.; Li, X.-B. MicroRNA-491-5p Suppresses Cervical Cancer Cell Growth by Targeting HTERT. Oncol. Rep. 2015, 34, 979–986.
- Huang, Y. Plk1 Upregulates Telomerase Activity by Affecting HTERT Stability. JBC 2015, 290, 18865–18873.
- Yalçin, Z.; Selenz, C.; Jacobs, J.J.L. Ubiquitination and SUMOylation in Telomere Maintenance and Dysfunction. Front. Genet. 2017, 8, 1–15.
- Lanna, A.; Coutavas, E.; Levati, L.; Rustin, M.H.A.; Henson, S.M.; Arne, N. IFN-α Inhibits Telomerase in Human CD8 + T Cells by Both HTERT Downregulation and Induction of P38 MAPK Signaling. J. Immunol. 2020, 191, 3744–3752.
- Nalobin, D.S.; Galiakberova, A.A.; Alipkina, S.I.; Glukhov, A.I. Regulation of Telomerase Activity. Biol. Bull. Rev. 2018, 8, 142–154.
- Macneil, D.E.; Bensoussan, H.J.; Autexier, C. Telomerase Regulation from Beginning to the End. Genes 2016, 7, 64.
- Singh, R.K.; Kumar, S.; Gautam, P.K.; Tomar, M.S.; Verma, P.K.; Singh, S.P.; Kumar, S.; Acharya, A. Protein Kinase C- α and the Regulation of Diverse Cell Responses. Biomol. Concepts 2017, 8, 143–153.
- Sui, J.; Zhang, S.; Chen, B.P.C. DNA–Dependent Protein Kinase in Telomere Maintenance and Protection. Cell. Mol. Biol. Lett. 2020, 25, 1–14.
- Abdelrahman, A.H.; Eid, M.M.; Hassan, M.; Eid, O.M.; AbdelKader, R.M.A.; AlAzhary, N.M.; Shahin, R.Y.; Sallam, M.T. Telomerase Reverse Transcriptase Gene Amplification in Hematological Malignancies. Egypt J. Med. Hum. Genet. 2019, 20, 1–9.
- Ghaffari, S.H.; Shayan-Asl, N.; Jamialahmadi, A.H.; Alimoghaddam, K.; Ghavamzadeh, A. Telomerase Activity and Telomere Length in Patients with Acute Promyelocytic Leukemia: Indicative of Proliferative Activity, Disease Progression, and Overall Survival. Ann. Oncol. 2008, 19, 1927–1934.
- Ishida, H.; Iguchi, A.; Aoe, M.; Takahashi, T.; Tamefusa, K.; Kanamitsu, K.; Fujiwara, K.; Washio, K.; Matsubara, T.; Tsukahara, H.; et al. Panel-Based next-Generation Sequencing Identifies Prognostic and Actionable Genes in Childhood Acute Lymphoblastic Leukemia and Is Suitable for Clinical Sequencing. Ann. Hematol. 2019, 98, 657–668.
- Alsultan, A.; Essa, M.; Aljefri, A.; Ayas, M.; Alharbi, M.; Alkhayat, N.; Al-Anzi, F.; Yassin, F.; Alkasim, F.; Alharbi, Q.; et al. Frequency of Pathogenic/Likely Pathogenic Germline Variants in Cancer-Related Genes among Children with Acute Leukemia in Saudi Arabia. Pediatr. Blood Cancer 2020, 67, e28340.
- Bruedigam, C.; Lane, S.W. Telomerase in Hematologic Malignancies. Curr. Opin. Hematol. 2016, 23, 346–353.
- Vicente-Dueñas, C.; Barajas-Diego, M.; Romero-Camarero, I.; González-Herrero, I.; Flores, T.; Sánchez-García, I. Essential Role for Telomerase in Chronic Myeloid Leukemia Induced by BCR-ABL in Mice. Oncotarget 2012, 3, 261–266.
- Röth, A.; Vercauteren, S.; Sutherland, H.J.; Lansdorp, P.M. Telomerase Is Limiting the Growth of Acute Myeloid Leukemia Cells. Leukemia 2003, 17, 2410–2417.
- Mohamad Ashari, Z.S.; Sulong, S.; Hassan, R.; Husin, A.; Sim, G.A.; Wahid, S.F.A. Low Level of TERC Gene Amplification between Chronic Myeloid Leukaemia Patients Resistant and Respond to Imatinib Mesylate Treatment. Asian Pac. J. Cancer Prev. 2014, 15, 1863–1869.
- Kawauchi, K. Activation of STAT5 Confers Imatinib Resistance on Leukemic Cells. Ph.D. Thesis, Department of Hematology, Tokyo Women’s Medical University, Tokyo, Japan, 2011.
- Augereau, A.; T’kint de Roodenbeke, C.; Simonet, T.; Bauwens, S.; Horard, B.; Callanan, M.; Leroux, D.; Jallades, L.; Salles, G.; Gilson, E.; et al. Telomeric Damage in Early Stage of Chronic Lymphocytic Leukemia Correlates with Shelterin Dysregulation. Blood 2011, 118, 1316–1322.
- Hoxha, M.; Fabris, S.; Agnelli, L.; Bollati, V.; Cutrona, G.; Matis, S.; Recchia, A.G.; Gentile, M.; Cortelezzi, A.; Morabito, F.; et al. Relevance of Telomere/Telomerase System Impairment in Early Stage Chronic Lymphocytic Leukemia. Genes Chromosomes Cancer 2014, 53, 612–621.
- Rondanin, R.; Simoni, D.; Romagnoli, R.; Baruchello, R.; Marchetti, P.; Costantini, C.; Fochi, S.; Padroni, G.; Grimaudo, S.; Pipitone, R.M. Inhibition of Activated STAT5 in Bcr/Abl Expressing Leukemia Cells with New Pimozide Derivatives. Bioorg. Med. Chem. Lett. 2014, 24, 4568–4574.
- Katsumi, S.; Kawauchi, K.; Ozaki, K.; Shimizu, S.; Kimura, T.; Motoji, T.; Yamada, O. Analysis of Molecular Mechanism Involved in Development of Acute Myeloid Leukemia. Gan Kagaku Ryoho Cancer Chemother. 2013, 40, 471–477.
- Schafranek, L.; Nievergall, E.; Powell, J.A.; Hiwase, D.K.; Leclercq, T.; Hughes, T.P.; White, D.L. Sustained Inhibition of STAT5, but Not JAK2, Is Essential for TKI-Induced Cell Death in Chronic Myeloid Leukemia. Leukemia 2015, 29, 76–85.
- Ghamlouch, H.; Nguyen-Khac, F.; Bernard, O.A. Chronic Lymphocytic Leukaemia Genomics and the Precision Medicine Era. Br. J. Haematol. 2017, 178, 852–870.
- Küppers, R.; Dalla-Favera, R. Mechanisms of Chromosomal Translocations in B Cell Lymphomas. Oncogene 2001, 20, 5580–5594.
- Nagel, I.; Szczepanowski, M.; Martín-Subero, J.I.; Harder, L.; Akasaka, T.; Ammerpohl, O.; Callet-Bauchu, E.; Gascoyne, R.D.; Gesk, S.; Horsman, D.; et al. Deregulation of the Telomerase Reverse Transcriptase (TERT) Gene by Chromosomal Translocations in B-Cell Malignancies. Blood 2010, 116, 1317–1320.
- Andreani, G.; Carrà, G.; Lingua, M.F.; Maffeo, B.; Brancaccio, M.; Taulli, R.; Morotti, A. Tumor Suppressors in Chronic Lymphocytic Leukemia: From Lost Partners to Active Targets. Cancers 2020, 12, 629.
- Rampazzo, E.; Bojnik, E.; Trentin, L.; Bonaldi, L.; Del Bianco, P.; Frezzato, F.; Visentin, A.; Facco, M.; Semenzato, G.; De Rossi, A. Role of MiR-15a/MiR-16-1 and the TP53 Axis in Regulating Telomerase Expression in Chronic Lymphocytic Leukemia. Haematologica 2017, 102, e253–e256.
- Hoffbrand, V.A.; Moss, P.A.H. Hoffbrand’s Essential Hematology, 7th ed.; Wiley Blackwell: Noida, India, October 2015.
- Lee, D.D.; Leão, R.; Komosa, M.; Gallo, M.; Zhang, C.H.; Lipman, T.; Remke, M.; Heidari, A.; Nunes, N.M.; Apolónio, J.D.; et al. DNA Hypermethylation within TERT Promoter Upregulates TERT Expression in Cancer. J. Clin. Investig. 2019, 129, 223–229.
- Zhao, X.; Tian, X.; Kajigaya, S.; Cantilena, C.R.; Strickland, S.; Savani, B.N.; Mohan, S.; Feng, X.; Keyvanfar, K.; Dunavin, N.; et al. Epigenetic Landscape of the TERT Promoter: A Potential Biomarker for High Risk AML/MDS. Br. J. Haematol. 2016, 175, 427–439.
- Staudt, D.; Murray, H.C.; McLachlan, T.; Alvaro, F.; Enjeti, A.K.; Verrills, N.M.; Dun, M.D. Targeting Oncogenic Signaling in Mutant FLT3 Acute Myeloid Leukemia: The Path to Least Resistance. Int. J. Mol. Sci. 2018, 19, 3198.
- Zhang, X.; Li, B.; Yu, J.; Dahlström, J.; Tran, A.N.; Björkhom, M.; Xu, D. MYC-Dependent Downregulation of Telomerase by FLT3 Inhibitors Is Required for Their Therapeutic Efficacy on Acute Myeloid Leukemia. Ann. Hematol. 2018, 97, 63–72.
- Osterwald, S.; Deeg, K.I.; Chung, I.; Parisotto, D.; Wörz, S.; Rohr, K.; Erfle, H.; Rippe, K. PML Induces Compaction, TRF2 Depletion and DNA Damage Signaling at Telomeres and Promotes Their Alternative Lengthening. J. Cell Sci. 2015, 128, 1887–1900.
- Zhang, X.; Li, B.; de Jonge, N.; Björkholm, M.; Xu, D. The DNA Methylation Inhibitor Induces Telomere Dysfunction and Apoptosis of Leukemia Cells That Is Attenuated by Telomerase Over-Expression. Oncotarget 2015, 6, 4888.
- Sandri, S.; De Sanctis, F.; Lamolinara, A.; Boschi, F.; Poffe, O.; Trovato, R.; Fiore, A.; Sartori, S.; Sbarbati, A.; Bondanza, A.; et al. Effective Control of Acute Myeloid Leukaemia and Acute Lymphoblastic Leukaemia Progression by Telomerase Specific Adoptive T-Cell Therapy. Oncotarget 2017, 8, 86987.
- Bashash, D.; Zareii, M.; Safaroghli-Azar, A.; Omrani, M.D.; Ghaffari, S.H. Inhibition of Telomerase Using BIBR1532 Enhances Doxorubicin-Induced Apoptosis in Pre-B Acute Lymphoblastic Leukemia Cells. Hematology 2017, 22, 330–340.