1. Introduction
Mitochondrial dysfunction, whether primary or secondary, is increasingly recognized as a hallmark of neurodegeneration
[1] and a wide body of literature provides evidence of impaired mitochondrial function as a cause rather than a consequence of neurodegeneration
[2][3][4][2,3,4]. Mitochondrial demise can be observed even before the appearance of pathognomonic histopathological hallmarks of the disease
[5]. Neurons are obligatorily dependent on mitochondrial energy production, which sustains a myriad of functions, including membrane remodeling, synaptic spine formation, and the generation of transmembrane resting and action potentials
[6][7][6,7].
Mitochondria also play a pivotal role in cell survival and death by regulating apoptotic pathways and contributing to different cellular functions including intracellular calcium homeostasis, maintenance of the cellular redox potential, and cell cycle regulation. Recent evidence demonstrated that mitochondria are also important in regulating cell fate towards stemness or neurogenesis
[8].
Thus, it is not surprising that mitochondrial dysfunction can have devastating effects on neuronal differentiation and survival. The brain is a major target in primary, genetically determined mitochondrial disease, but mitochondrial dysfunction is also a prominent feature in many of the most prevalent neurodegenerative diseases, including Parkinson’s Disease (PD)
[9], Huntington’s Disease
[10], neurodegeneration associated with stroke
[11], Amyotrophic Lateral Sclerosis
[12], neurodegenerative ataxias
[13] and different types of psychiatric and cognitive disorders such as Dementia with Lewy Bodies
[14], Frontotemporal Dementia
[15], and Alzheimer’s disease (AD)
[16]. In AD, for example, the “mitochondrial cascade” hypothesis proposes that organellar dysfunction is the primary event in AD pathology
[17]. Moreover, although the extracellular deposition of amyloid-beta (Aβ) peptides (Aβ
1–40, Aβ
1–42) is the key histopathological hallmark of AD, Aβ accumulation is proposed to also occur in mitochondria, through the mitochondrial import machinery
[18][19][18,19], causing impairment of different mitochondrial pathways such as respiration, reactive oxygen species (ROS) detoxification, and organelle morphology
[20][21][22][23][24][25][26][20,21,22,23,24,25,26].
The protease-mediated quality control system is a first-line homeostatic defense against mitochondrial damage, and includes degradation of non-assembled, misfolded or damaged proteins as a result of oxidative stress, elimination of cleaved products during protein processing, and overall regulation of protein turnover and homeostasis (referred to as proteostasis)
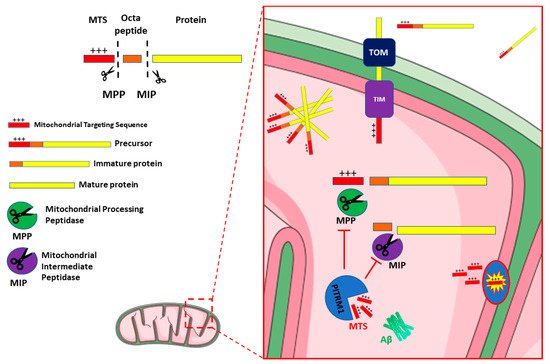
[27]. The proteolytic system in mitochondria is crucial for the maintenance of protein turnover and the integrity of mitochondria. The majority of mitochondrial proteins are synthesized on cytosolic ribosomes with an N-terminal peptide (the presequence or mitochondrial targeting sequence—MTS) which is recognized by—and binds to—specific receptors in the mitochondrial outer membrane. Following this event, these precursor mitochondrial proteins are translocated through the mitochondrial entry gate, the TOM (Translocase of Outer Membrane) complex and then, via a specific TIM (Translocase of Inner Membrane) system, TIM23, into the matrix
[28], where they undergo proteolytic processes, including the cleavage of MTS, and structural modifications that lead to mature, functional proteins
[29].
Several proteases and peptidases have been identified in different mitochondrial sub compartments. Most are ATP dependent proteases, such as the matrix-located Lon protease 1 (LONP1) and the membrane-bound AAA (ATPases Associated with diverse cellular Activities) proteases. The latter include i-AAA (Yme1), active in the intermembrane space (IMS), and m-AAA (Yta10/Yta12), exposed to the matrix. These enzymes catalyse the initial step of degradation, cleaving proteins into peptides, thus contributing to mitochondrial quality control
[30]. Other ATP-independent proteases such as the mitochondrial processing peptidase (MPP), the mitochondrial intermediate protease (MIP), and the inner membrane peptidase (IMP), also generate short peptides. Upon import of the mitochondrial precursor proteins, MPP in the matrix cleaves the MTSs, releasing the mature proteins
[31][32][33][31,32,33]. MPP activity can produce import intermediates, which are further processed by MIP (the octapeptidyl peptidase Oct1 in yeast) or IMP (the intermediate cleaving peptidase Icp55 in yeast)
[33][34][35][36][33,34,35,36]. Therefore, all these proteases are involved in the processing of precursor protein and release free MTS peptides after proteolytic cleavage (
Figure 1)
[37].
Figure 1. Schematic representation of PITRM1 function and interaction. Mitochondrial Precursor proteins are imported from cytosol to the matrix through the Translocase of the Outer Membrane (TOM) and Translocase of the Inner Membrane (TIM) complexes. Dysfunctional activity of PITRM1 results in the accumulation of Amyloid beta (Aβ), of Mitochondrial Targeting Sequences (MTS) and of octapeptides that trigger feedback inhibition of Mitochondrial Processing Peptidase (MPP) and Mitochondrial Intermediate Peptidase (MIP), leading to the accumulation and aggregation of unprocessed precursor.
Figure 1 was modified from SMART (Servier Medical Art), licensed under a Creative Common Attribution 3.0 Generic License.
http://smart.servier.com/.
Typically, the MTSs are amphiphilic species, with a polar, positively charged, arginine-rich side, opposite to an apolar side (
Figure 1). Therefore, if these MTS peptides fail to be cleared from the mitochondrial matrix, they may act as detergent-like, toxic agents, forming pores in the membranes and resulting in uncoupling of oxidative phosphorylation and dissipation of the mitochondrial membrane potential
[36].
2. PITRM1/PreP
PITRM1, also termed Presequence protease (PreP), is localized in the mitochondrial matrix; it is the only known protein responsible for the degradation of the MTS, thus completing the last step of the protein import process. PreP was initially identified in
Arabidopsis thaliana (AtPreP), and shown to degrade the MTS of both mitochondria and chloroplasts
[38]. A yeast mutant lacking the
PITRM1 homolog
Cym1 [39] displayed mitochondrial accumulation of precursor proteins and processing intermediates, as well as decreased levels of cleaved, mature proteins
[32][40][32,40]. Impaired preprotein maturation leads to accelerated protein degradation and an unbalanced mitochondrial proteome, resulting in mitochondrial dysfunction manifested by reduced respiration, altered membrane potential, and high ROS levels
[28][37][28,37].
The human gene PITRM1 is located in chromosomal region 10p15.2, contains 27 exons, and is transcribed from the antisense strand. Three different isoforms of the protein correspond to transcript variants derived from alternative splicing. In isoforms 1 and 2, all 27 exons of the gene are retained and translated, with a total of 1038 and 1037 aminoacids (aa), respectively, due to the use of two different splice donor sites at exon 17. The third isoform is the smallest one and contains 939 aa derived from 24 exons of the gene.
Human PITRM1 was initially identified as metalloprotease 1
[41], and shows 31% sequence identity to AtPreP, performing a similar function in human mitochondria. PITRM1 belongs to the M16 metalloproteases family, which are Zn
2+-dependent and ATP-independent enzymes that share a conserved architecture of two ~ 50 kDa homologous domains enclosing a large catalytic chamber. Three families, M16-A, -B and -C, have been characterized depending on the connection between these two domains
[42]. PITRM1 is a 117 kDa M16C enzyme arranged in four domains, forming two enzyme halves, hPreP-N (amino acids 33–509) and hPreP-C (aa 576–1037) domains, connected by a hinge region (aa 510–575), which is possibly involved in the opening and closing of the two enzyme halves. Active site residues are located in both halves, which interact to form a large peptidasome chamber
[43]. In addition to targeting peptides generated during the import processing, the large catalytic chamber of PITRM1 is also able to degrade a wide range of unstructured peptides, ranging from 10 to 65 aa, but not larger folded proteins
[44].
In vitro studies demonstrated that human PITRM1 can degrade Aβ
1–40, Aβ
1–42 and the Aβ arctic (Aβ
1–40 E22G), a peptide that causes increased fibril formation and early onset of a familial variant of AD. Its proteolytic activity generates several fragments, which are unique to PITRM1 (in comparison to other proteases), and recognizes the cleavage sites in the very hydrophobic C-terminal portion of Aβ that is prone to aggregation
[38]. Interestingly, the 3D structures of PITRM1 are highly similar to IDE (Insulin degrading enzyme), a zinc metallopeptidase that degrades intracellular insulin, as well as glucagon, amylin, beta-amyloid, bradykinin, and kallidin. Moreover, IDE has been suggested to have a role in the degradation of cleaved MTSs
[37][45][37,45]. The preferential affinity of this enzyme for insulin results in the insulin-mediated inhibition of degradation of other peptides, such as beta-amyloid
[46]. Variants in the
IDE gene and deficiencies in the protein function have previously been associated with type 2 diabetes mellitus and Alzheimer’s disease
[47][48][47,48].