BA affect the functioning of the receptors for several neurotransmitters, including M
2 and M
3 muscarinic acetylcholine receptors, γ-aminobutyric acid (GABA) type A (GABA
A) receptors, and N-methyl-D-aspartate (NMDA) receptors. TCA, GDCA, and TDCA activate the M
2 receptor, while DCA activates the M
3 receptor. M
2 receptors are distributed throughout the brain and are crucial for cognitive function. However, M
3 receptors are localized to neurons, which project to regions such as the hippocampus and substantia nigra, although they are also expressed at low levels throughout the brain. Thus, BA may affect cognitive function, memory, and learning
[4][140]. NMDA receptors are ionotropic glutamate receptors that are activated by the simultaneous binding of both glutamate and D-serine or glycine
[5][6][141,142]. Their activation causes Ca
2+ influx, which can induce long-term potentiation and long-term depression. Therefore, appropriate NMDA receptor activation is important for learning and memory
[7][143]. In contrast, the GABA
A receptor is an ionotropic GABA receptor that is a ligand-gated chloride ion channel. The activation of GABA
A receptor causes an influx of chloride ions, leading to the hyperpolarization of neurons and the inhibition of neurotransmission
[8][144]. CDCA, DCA, and CA block both GABA
A and NMDA receptors
[9][145]. Histaminergic neurons in the tuberomammillary nucleus (TMN) of the hypothalamus play an important role in arousal and wakefulness
[10][146] and express GABA
A receptors; thus, the suppression of histaminergic neuronal activation in the TMN by GABA induces sleep
[11][147]. Conversely, UDCA increases arousal by blocking GABA
A receptors on TMN neurons
[12][148]. Moreover, a recent study has demonstrated that TUDCA can induce neurogenesis in adult rats
[13][149]. Adult neurogenesis is the process of generating new functional neurons that are added to the adult brain and occurs in two specific regions: The subgranular zone (SGZ) of the dentate gyrus (DG) in the hippocampus and the subventricular zone (SVZ), located in the walls of the lateral ventricles. Neural stem cells (NSCs) in the SGZ of adult mammals generate neurons in the DG. NSCs in adult human SVZ may produce functional neurons in the striatum by migrating there. In contrast, NSCs in adult rodent SVZ may produce functional neurons in the olfactory bulb
[14][150]. In the rat, TUDCA increases the proliferation and neural differentiation of NSCs in the SVZ, but not in the DG
[13][149].
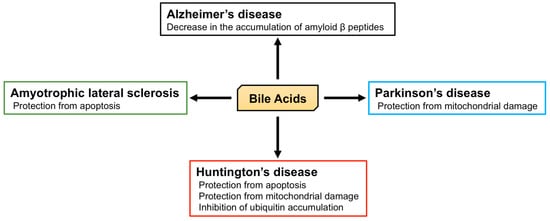
2.2. The Role of BA in Neurodegenerative Diseases
Alzheimer’s disease is characterized by memory loss, dementia, and morphological changes in the brain and is a common progressive neurodegenerative disease. The main pathological feature of the brains of patients with Alzheimer’s disease is the accumulation of amyloid β peptides and tangles of tau protein
[15][151]. The processing of amyloid precursor protein (APP) by β and γ secretases generates amyloid β peptide
[16][17][152,153] and the γ secretase complex includes presenilin 1 (PS1), which is associated with the maturation of V-ATPase, responsible for the acidification of lysosomes. PS1 dysfunction, thus, leads to an impairment in lysosomal acidification and function
[18][19][154,155]. The accumulation of amyloid β peptides is relevant to the dysfunction of both APP and γ secretase, and APP/PS1 double knockout mice, which express both mutated human APP and PS1, are used as a model of Alzheimer’s disease. Interestingly, TUDCA reduces the accumulation of amyloid β peptides in the hippocampus and frontal cortex and rescues memory deficits in APP/PS1 double knockout mice
[20][21][156,157]. In humans, plasma CA concentrations in patients with Alzheimer’s disease are significantly lower than those in control subjects, and the TCA concentration in the brain of patients with Alzheimer’s disease is also significantly lower
[22][42]. In contrast, the plasma concentration of LCA, a secondary BA, is significantly higher in patients with Alzheimer’s disease than in controls
[23][158]. Furthermore, it has been shown that the ratio of DCA (secondary BA) to CA (primary BA) in serum is significantly higher in Alzheimer’s patients
[24][159]. These studies indicate the relationship between BA and Alzheimer’s disease, and the importance of the brain-gut-microbiome axis. However, morphological and functional abnormalities in mitochondria have also been identified in patients with Alzheimer’s disease
[25][26][160,161]. In fibroblasts from Alzheimer’s disease patients, the mitochondrial membrane potential (MMP) is lower, and there is mitochondrial elongation. Dynamin-related protein 1 (DRP1) plays an important role in mitochondrial fission and is essential for mitochondrial quality control
[27][162]. Although the expression of DRP1 is lower in fibroblasts from Alzheimer’s disease patients, UDCA increases its expression
[28][163].
Parkinson’s disease is another common progressive neurodegenerative disease that is characterized by tremors, muscle stiffness, loss or impairment of voluntary movements, slowness of movement, and postural instability
[29][164]. Mutations in phosphatase and tensin homolog-induced putative kinase 1 (PINK1) and parkin are found in early-onset Parkinson’s disease and are important in the progression of Parkinsonism
[30][31][32][33][165,166,167,168]. PINK1 and parkin initiate mitophagy and nuclear dot protein 52 kDa (NDP52), and optineurin are essential for this process
[34][35][169,170]. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and rotenone treatment have been widely used in the creation of a model of Parkinson’s disease. Both toxins inhibit complex I in mitochondria, leading to neuronal mitochondrial damage and Parkinsonism
[36][37][171,172]. However, TUDCA protects neurons from MPTP-induced oxidative stress and neurotoxicity in the midbrain and striatum of mice
[38][173]. TUDCA induces nuclear factor erythroid 2-related factor 2 (Nrf2) expression and reduces the generation of reactive oxygen species, and indeed, this anti-oxidant effect of TUDCA may be Nrf2-dependent
[39][174]. Moreover, TUDCA ameliorates clinical signs induced by MPTP, such as the increase in swimming latency, foot-dragging, and tremors. In addition, TUDCA inhibits the loss of dopaminergic neurons and reduces the loss of MMP and mitochondrial mass elicited by MPTP
[40][175]. Furthermore, TUDCA increases the expression of PINK1, parkin, and the ratio of LC3-II/lC3-I, implying that TUDCA induces mitophagy
[41][176]. In addition, in the rotenone-induced rat model of Parkinson’s disease, UDCA ameliorates abnormalities in the mitochondria of striatal neurons, such as irregular swelling and loss of cristae
[42][177].
Huntington’s disease is an autosomal dominant inherited neurodegenerative disease caused by a CAG trinucleotide expansion encoding polyglutamine (polyQ) at the N-terminus of Huntingtin (HTT). Huntington’s disease is characterized by motor dysfunction, cognitive decline, and psychiatric disturbances
[43][44][178,179]. Chemical and genetic models of Huntington’s disease have been studied
[45][46][180,181]. Firstly, 3-nitropropionic acid (3-NP) is an inhibitor of succinate dehydrogenase (complex II) in mitochondria and induces degeneration of the caudate-putamen, which is also present in Huntington’s disease
[45][180]. 3-NP treatment is associated with swelling of striatal mitochondria, abnormal mitochondrial membrane structure, and apoptosis. However, TUDCA prevents mitochondrial damage and apoptosis and ameliorates the sensorimotor deficits induced by 3-NP
[47][182]. Secondly, R6/2 transgenic mice possess a genomic fragment containing exon 1 of the human
Huntingtin gene, which carries 144 CAG repeats, and are the most widely used model of Huntington’s disease
[46][48][181,183]. In these mice, TUDCA prevents the striatal apoptosis and cerebral and striatal atrophy
[49][184]. The accumulation of HTT and ubiquitin are also symptoms of Huntington’s disease, and ubiquitin is recruited to polyglutamine-expanded HTT fragments
[50][185]. TUDCA reduces the accumulation of ubiquitin in the striatum of R6/2 transgenic mice and ameliorates their sensorimotor deficits
[49][184].
Amyotrophic lateral sclerosis (ALS) is a progressive and ultimately fatal disease that is characterized by the degeneration of both upper and lower motor neurons, leading to muscle weakness, atrophy, and paralysis
[51][186]. Impairments in superoxide dismutase 1 (SOD1), C9ORF72, TAR DNA-binding protein of 43 kDa (TDP-43), and fused in sarcoma (FUS) are molecular features of this disease
[52][187]. Transgenic mice that carry the glycine 93 to alanine mutation of human SOD1 (hSOD1
G93A) are the most studied model of ALS
[53][188]. NSC-34 cells carrying hSOD1
G93A also represent a useful model of ALS-affected motor neurons
[54][189]. Glycoursodeoxycholic acid (GUDCA) prevents the apoptosis of NSC-34 cells carrying hSOD1
G93A [55][190], and clinical trials of the use of TUDCA in patients with ALS have shown an improvement in muscle function and survival time, without adverse effects
[56][191].