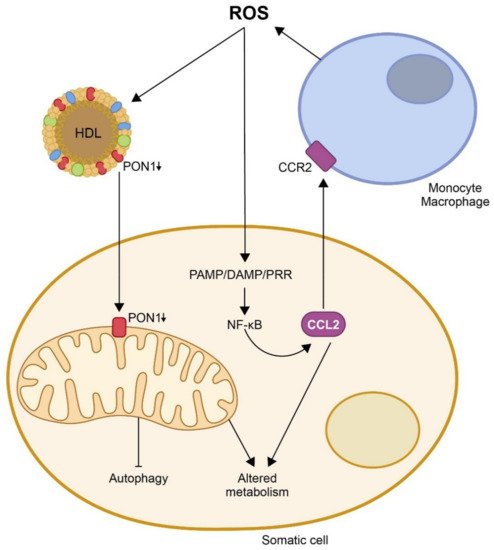
One of the proposed mechanisms by which oxidative stress would enhance the inflammatory response is the induction and assembly of multiprotein complexes called inflammasomes. ROS activate the NOD-like receptor Pyrin domain 3 (NLRP3) in macrophages, triggering the formation of inflammasomes and an immune reaction that involves the synthesis of pro-inflammatory chemokines, from which CCL2 is, probably, the most representative
[28][29][30][31][58,59,60,61]. This chemokine is upregulated following tissue injury and is expressed by both inflammatory and stromal cells. CCL2 has been reported to promote endoplasmic reticulum (ER) stress and autophagy and to regulate NF-ĸB expression by catalyzing de-ubiquitination
[32][62]. The main pathway triggering inflammation is, probably, the activation of pattern-recognition receptors (PRRs), which recognize pathogen-associated molecular patterns (PAMPs), synthesized as a response to pathogens, or damage-associated molecular patterns (DAMPs), which are products of damaged cells
[33][34][35][63,64,65]. The binding of PAMP/DAMP to a PRR leads to NF-ĸB activation and the production of adhesion molecules and chemokines that lead to infiltration of immune cells into the sites of tissue damage
[36][66]. Other alternative pathways also result in similar outcomes, particularly the phosphoinositide 3-kinase-related signaling pathway, the mitogen-activated protein kinase pathway, and the Janus kinase/signal transducers and activators of transcription signaling pathway
[37][38][39][67,68,69]. These changes induce the UPR, essentially by three ER-related transmembrane proteins, i.e., the inositol-requiring enzyme 1, the protein kinase RNA-like endoplasmic reticulum kinase, and the activating transcription factor 6
[40][41][42][43][70,71,72,73]. CCL2 and other chemokines, together with oxidative stress, trigger ER stress. In addition, the UPR may regulate inflammation through several pathways, such as the regulation of oxidative stress or the upregulation of CCR2 expression
[44][74]; the UPR links ER stress with cell death and autophagy
[45][75]. When cell damage is moderate, autophagy helps cells survive the injury, allowing them to heal and thus preventing cell death by removing toxic protein aggregates. However, when cell damage is high, the result is a non-apoptotic form of cell death that can be detrimental. The role of autophagy in the maintenance of mitochondrial integrity seems to be paramount
[46][76]. Mitophagy increases cell lifespan, while repression of autophagy reduces lifespan. Several studies have linked mitochondrial dysfunction, autophagy, and age-related diseases with the activity of the inflammasomes
[47][48][49][50][77,78,79,80]. Taken together, these results define a clear relationship between oxidative stress, chemokines, and mitochondrial impairment, resulting in metabolic alterations and their involvement in diseases.
Activation of the immune response and chronic inflammation has been associated with aging and age-related diseases
[51][52][53][81,82,83]. Senescent cells secrete chemokines, which influence the trafficking of immune cells
[54][55][84,85]. Epidemiological studies have suggested that CCL2 levels are increased in older individuals, independently of metabolic alterations. Moreover, in vitro studies have shown that chemokines appear to confer senescence to neighboring normal cells in an autocrine and paracrine fashion
[56][57][58][86,87,88]. A recent study by our research group in mice with accelerated aging is a good example of such relationships
[59][89]. We crossbred mice that overexpressed CCL2 with progeroid mice bearing a mutation in the
lamin A gene. Wild-type animals and progeroid mice not overexpressing CCL2 were used as controls. We observed that progeroid mice lost weight (relative to the wild-type animals) and developed lordokyphosis and lipodystrophy. The lifespan was significantly reduced in both strains of progeroid mice, but this reduction was higher in those overexpressing CCL2. These mice also presented specific characteristics of metabolic dysregulation in skeletal muscle, including alterations in the glucose and TCA cycles, and in one-carbon metabolism. These data suggest that mitochondrial metabolites play major roles in pathological aging. Consequently, we investigated the mitochondrial respiratory complexes in skeletal muscle, and observed that the expressions of complexes I and V were lower in mice overexpressing CCL2. In addition, the protein concentrations of translocase of outer membrane 20 (TOM20) and mitofusin 2 (MFN2) in the muscles of the progeroid mice were decreased, indicating alterations in the correct formation of the mitochondrial network. We also observed an increase in p53, which would indicate the triggering of aging through a p53-mediated transcriptional program involving the mechanistic target of rapamycin. Indeed, we found inhibition of phosphorylation of phosphoinositide 3-kinase, indicating a mechanistic target of rapamycin inhibition. Finally, the higher microtubule-associated proteins 1A/1B light chain 3B (LC3) II/I ratio, and lower lysosome-associated membrane protein 2 (LAMP2A) and sequestosome 1 (p62) expressions suggested the involvement of chaperone-mediated autophagy as a consequence of the CCL2 overexpression in the progeroid animals.
Following on from the actions described above, in many infectious and non-infectious diseases, the adaptive immunity deteriorates, whereas the innate immunity is more responsive to stimuli; the consequence is the development of an inflammatory reaction. Several studies have linked the activation of NLRP3, which is dependent on increased generation of free radical species by mitochondria, with metabolic disturbances
[60][61][62][90,91,92]. Hence, it is of note that awareness of the origin of free radicals and the putative mechanisms of prevention (i.e., PON1) is critical when establishing possible therapeutic interventions in order to preempt an inflammatory reaction. Within this context, the interaction of PON1 with CCL2 can play a key role.