The recent formulation, production, and ongoing administration of vaccines represent a starting point in the battle against SARS-CoV-2, but they cannot be the only aid available. In this regard, the use of drugs capable to mitigate and fight the virus is a crucial aspect of the pharmacological strategy. Among the plethora of approved drugs, a consistent element is a heterocyclic framework inside its skeleton. Heterocycles have played a pivotal role for decades in the pharmaceutical industry due to their high bioactivity derived from anticancer, antiviral, and anti-inflammatory capabilities. In this context, the development of new performing and sustainable synthetic strategies to obtain heterocyclic molecules has become a key focus of scientists.
- antiviral
- heterocyclization
- metal-promoted
1. Introduction
Heterocyclic compounds have versatile applications across many chemistry fields. N, S, and O are the most common heteroatoms, and their corresponding heterocycles can be found as the main structural units in synthetic pharmaceuticals and agrochemicals, as well as widely present in nature in plant alkaloids, nucleic acids, anthocyanins, and flavones [19][1]. Drugs containing a heterocyclic moiety inside their structure show antitumor, anti-inflammatory, antifungal, antidepressant, anti-HIV, antimalarial, and antiviral properties [20,21,22][2][3][4]. In particular, the latter three properties are central in the fight against SARS-CoV-2 [23,24,25][5][6][7]. Over the years, due to the importance of these small molecules, synthetic organic chemists have focused their efforts on the development of synthetic protocols which are more and more efficient, atom-economical, and environmentally friendly. Metal-catalyzed protocols, involving all metals from transition to rare-earth metals, have attracted the attention of chemists as compared to other synthetic methodologies because they directly employ easily available substrates to build multi-substituted complex molecules under mild conditions. Metal-catalyzed heterocyclization starting from acyclic precursors is considered a very performant tool in drug synthesis [26][8]. In this review, we focus our attention on metal-catalyzed heterocyclization methodologies for achieving pivotal scaffolds associated with molecules showing anti-COVID-19 properties.
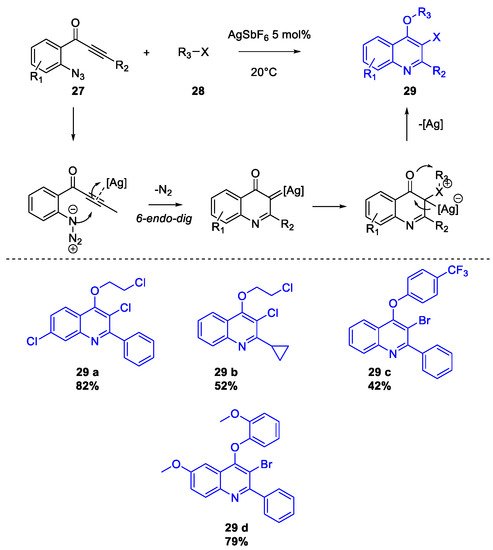
2. Chloroquine and Hydroxychloroquine
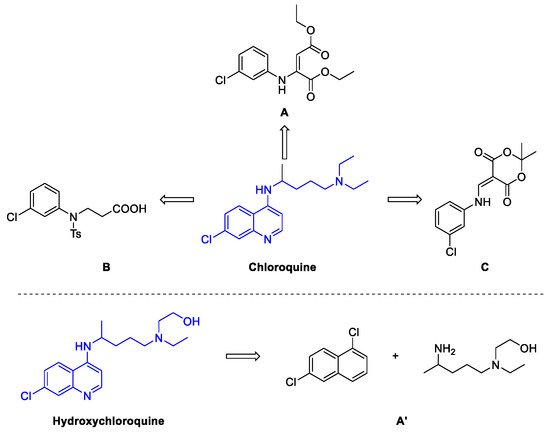
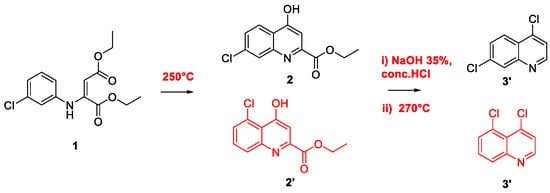
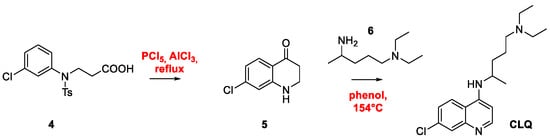
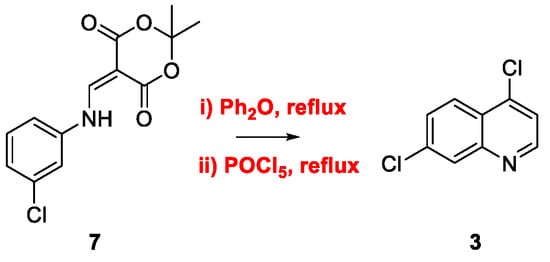
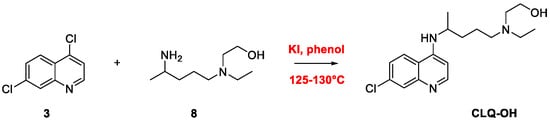
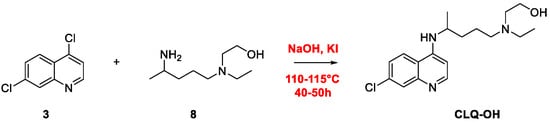
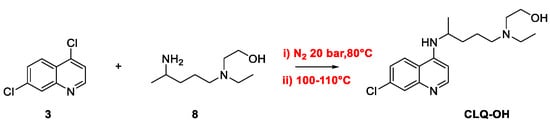
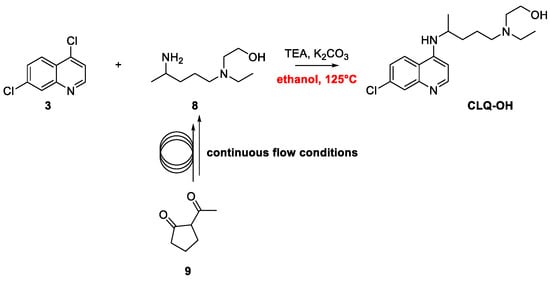
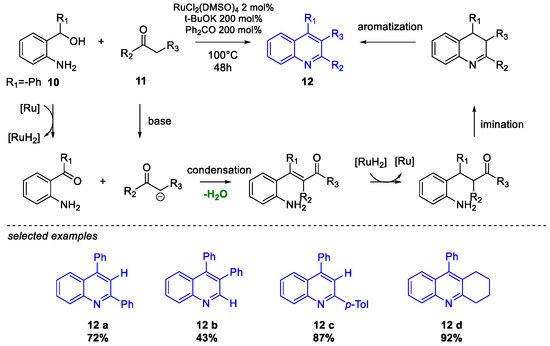
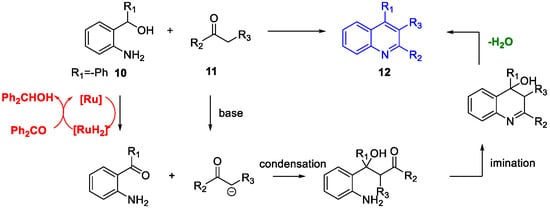
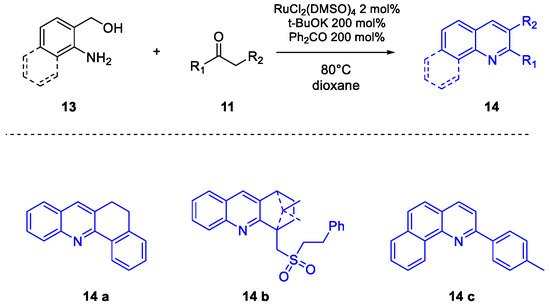
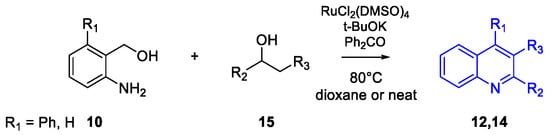
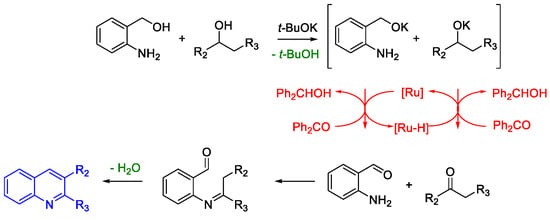
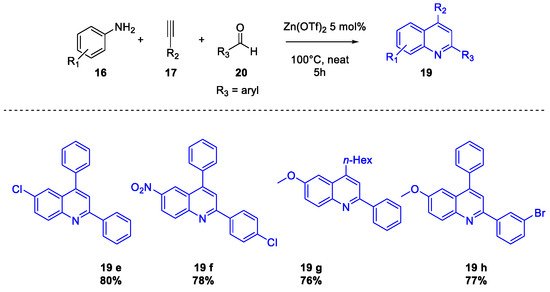
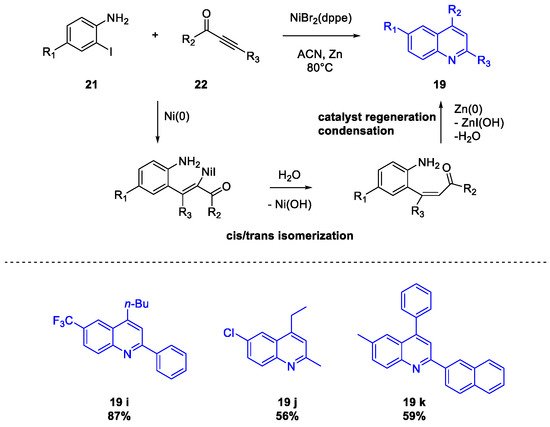
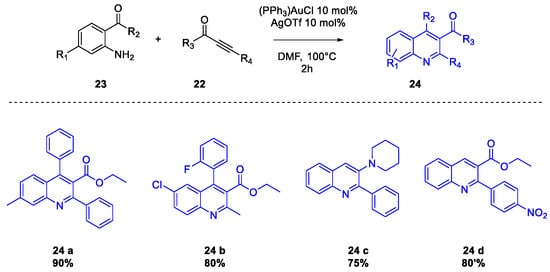
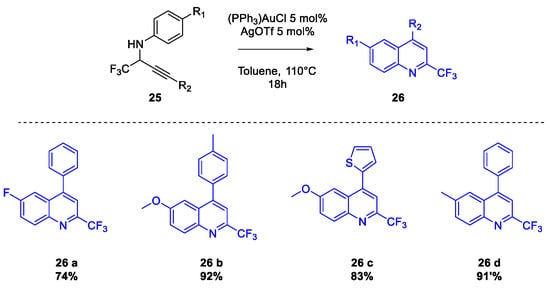
Quinoline Synthesis: Metal-Promoted Annulation
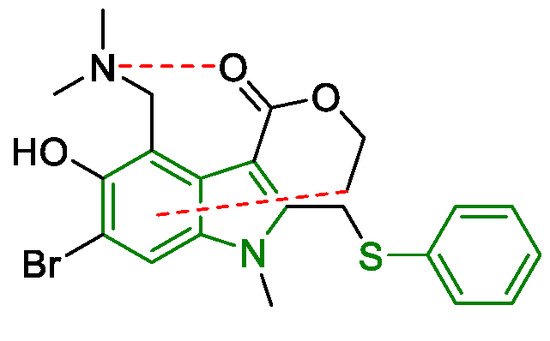
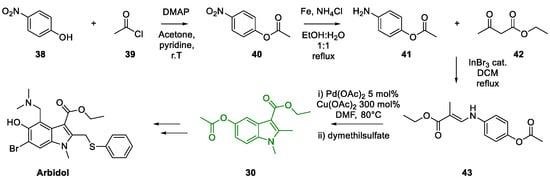
3. Arbidol
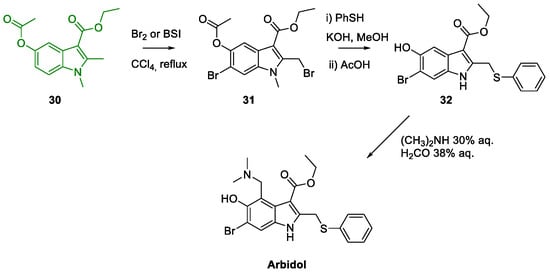
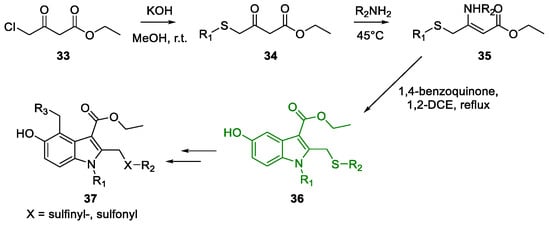
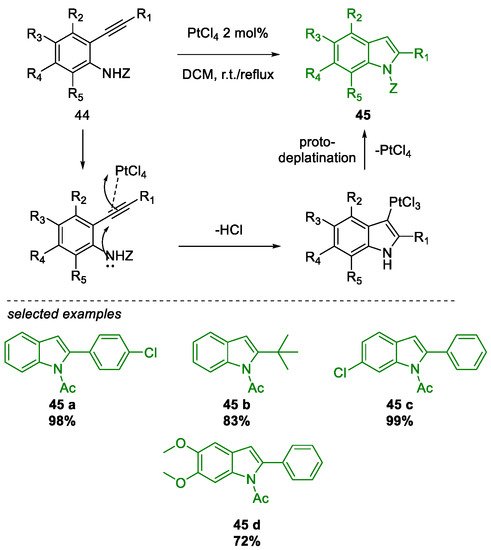
Metal-Promoted Heterocyclization to Achieve Polysubstituted Indoles
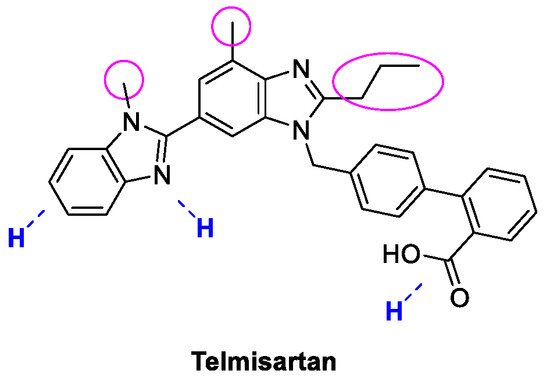
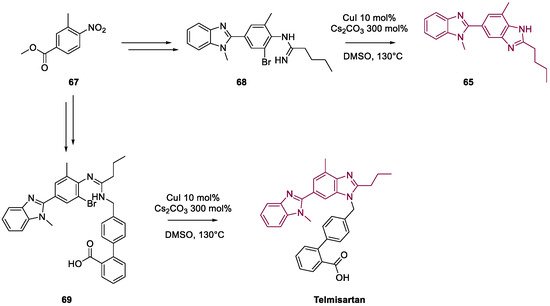
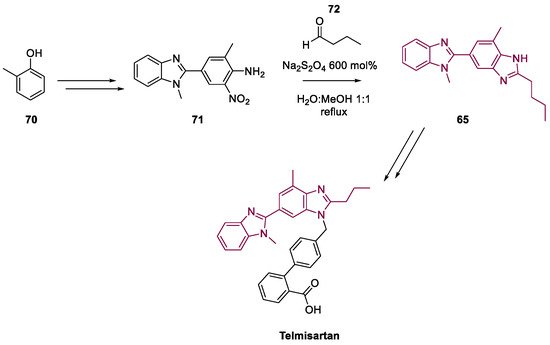
4. Telmisartan
5. Quercetin and Luteolin
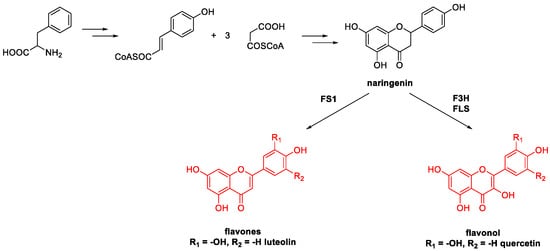
Metal-Catalyzed O-Heterocyclization to Flavonoids
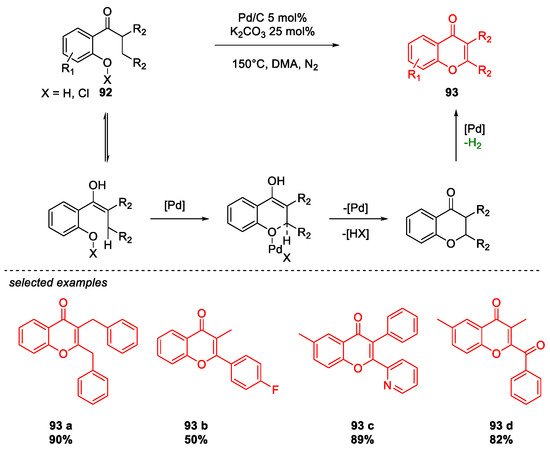
6. SARS-CoV-2 3CL Protease Target Drugs
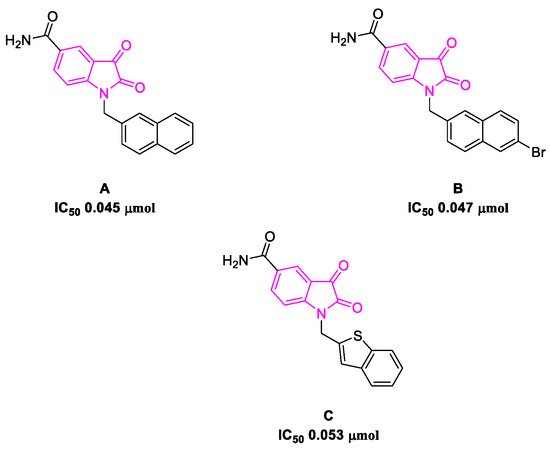
The SARS-CoV-2 3C-like protease is the main protease present in the virus, and it is crucial in the translation process from polyproteins to viral RNA [107][55]. It was demonstrated that the catalytic domain (Cys-145 and His-41) is particularly conserved, which makes the 3CL protease an attractive target for broad-spectrum anti-coronavirus therapies and drug discovery [108][56]. The SARS-CoV-2 main protease and spike protein are essential for the transmission of the virus and the severity of the infection in the host. Suppressing one or both biological targets can address the concerns linked to transmission, whereby acute COVID-19 symptoms can be drastically minimized [109][57]. Potential 3CL protease inhibitors reported in the literature have been screened to test their efficacy. Among the prospective bioactive molecules targeting this protein, ritonavir in combination with lopinavir and N-decorated isatins has shown promising results in the fight against SARS-CoV-2 [16,110,111][58][59][60].
7. Conclusions
References
- Pathan, S.I.; Chundawat, N.S.; Chauhan, N.P.S.; Singh, G.P. A review on synthetic approaches of heterocycles via insertion-cyclization reaction. Synth. Commun. 2020, 50, 1251–1285.
- Negi, M.; Chawla, P.A.; Faruk, A.; Chawla, V. Role of heterocyclic compounds in SARS and SARS CoV-2 pandemic. Bioorg. Chem. 2020, 104, 104315.
- Hagar, M.; Ahmed, H.A.; Aljohani, G.; Alhaddad, O.A. Investigation of Some Antiviral N-Heterocycles as COVID 19 Drug: Molecular Docking and DFT Calculations. Int. J. Mol. Sci. 2020, 21, 3922.
- Gomtsyan, A. Heterocycles in drugs and drug discovery. Chem. Heterocycl. Compd. 2012, 48, 7–10.
- Das, R.R.; Jaiswal, N.; Dev, N.; Jaiswal, N.; Naik, S.S.; Sankar, J. Efficacy and Safety of Anti-malarial Drugs (Chloroquine and Hydroxy-Chloroquine) in Treatment of COVID-19 Infection: A Systematic Review and Meta-Analysis. Front. Med. 2020, 7.
- Sang, P.; Tian, S.-H.; Meng, Z.-H.; Yang, L.-Q. Anti-HIV drug repurposing against SARS-CoV-2. RSC Adv. 2020, 10, 15775–15783.
- Stebbing, J.; Phelan, A.; Griffin, I.; Tucker, C.; Oechsle, O.; Smith, D.; Richardson, P. COVID-19: Combining antiviral and anti-inflammatory treatments. Lancet Infect. Dis. 2020, 20, 400–402.
- Santhoshkumar, R.; Cheng, C. Reaching Green: Heterocycle Synthesis by Transition Metal-Catalyzed C−H Functionalization in Sustainable Medium. Chem. Eur. J. 2019, 25, 9366–9384.
- Li, D.; Hu, J.; Li, D.; Yang, W.; Yin, S.-F.; Qiu, R. Reviews on Biological Activity, Clinical Trial and Synthesis Progress of Small Molecules for the Treatment of COVID-19. Top. Curr. Chem. 2021, 379, 4.
- Kapoor, K.M.; Kapoor, A. Role of chloroquine and hydroxychloroquine in the treatment of COVID-19 infection—A systematic literature review. medRxiv 2020.
- Meyerowitz, E.A.; Vannier, A.G.L.; Friesen, M.G.N.; Schoenfeld, S.; Gelfand, J.A.; Callahan, M.V.; Kim, A.Y.; Reeves, P.M.; Poznansky, M.C. Rethinking the role of hydroxychloroquine in the treatment of COVID-19. FASEB J. 2020, 34, 6027–6037.
- Pagliano, P.; Piazza, O.; De Caro, F.; Ascione, T.; Filippelli, A. Is Hydroxychloroquine a Possible Postexposure Prophylaxis Drug to Limit the Transmission to Healthcare Workers Exposed to Coronavirus Disease 2019? Clin. Infect. Dis. 2020, 71, 887–888.
- Patil, V.M.; Singhal, S.; Masand, N. A systematic review on use of aminoquinolines for the therapeutic management of COVID-19: Efficacy, safety and clinical trials. Life Sci. 2020, 254, 117775.
- Surrey, A.R.; Hammer, H.F. Some 7-Substituted 4-Aminoquinoline Derivatives. J. Am. Chem. Soc. 1946, 68, 113–116.
- Johnson, W.S.; Buell, B.G. A New Synthesis of Chloroquine. J. Am. Chem. Soc. 1952, 74, 4513–4516.
- Margolis, B.J.; Long, K.A.; Laird, D.L.T.; Ruble, J.C.; Pulley, S.R. Assembly of 4-Aminoquinolines via Palladium Catalysis: A Mild and Convenient Alternative to S N Ar Methodology. J. Org. Chem. 2007, 72, 2232–2235.
- Surrey, A.R.; Hammer, H.F. The Preparation of 7-Chloro-4-(4-(N-ethyl-N-β-hydroxyethylamino)-1-methylbutylamino)-quinoline and Related Compounds. J. Am. Chem. Soc. 1950, 72, 1814–1815.
- Kumar, A.V.; Vyas, K.D.; Singh, D.; Nanolavadekar, S.; Bhiae, S.; Jadhav, A. An Improved Process for the Preparation of 7-chloro-4-(5-N-Ethyl-N-2-Hydroxyethylamine)-2-pentyl Aminoquinoline and Its Intermediates. U.S. Patent WO 2005062723, 11 July 2005.
- Min, Y.S.; Cho, H.S.; Mo, K.W. New Preparation of Hydroxychloroquine. U.S. Patent WO 2010027150, 17 March 2010.
- Yu, E.; Mangunuru, H.P.R.; Telang, N.S.; Kong, C.J.; Verghese, J.; Gilliland III, S.E.; Ahmad, S.; Dominey, R.N.; Gupton, B.F. High-yielding continuous-flow synthesis of antimalarial drug hydroxychloroquine. Beilstein J. Org. Chem. 2018, 14, 583–592.
- Chelucci, G.; Porcheddu, A. Synthesis of Quinolines via a Metal-Catalyzed Dehydrogenative N-Heterocyclization. Chem. Rec. 2017, 17, 200–216.
- Sharma, R.; Kour, P.; Kumar, A. A review on transition-metal mediated synthesis of quinolines. J. Chem. Sci. 2018, 130, 73.
- Eswaran, S.; Adhikari, A.V.; Chowdhury, I.H.; Pal, N.K.; Thomas, K.D. New quinoline derivatives: Synthesis and investigation of antibacterial and antituberculosis properties. Eur. J. Med. Chem. 2010, 45, 3374–3383.
- Ramann, G.; Cowen, B. Recent Advances in Metal-Free Quinoline Synthesis. Molecules 2016, 21, 986.
- Matada, B.S.; Yernale, N.G. The contemporary synthetic recipes to access versatile quinoline heterocycles. Synth. Commun. 2021, 51, 1133–1159.
- Martínez, R.; Ramón, D.J.; Yus, M. RuCl2(dmso)4 Catalyzes the Solvent-Free Indirect Friedländer Synthesis of Polysubstituted Quinolines from Alcohols. European J. Org. Chem. 2007, 2007, 1599–1605.
- Martínez, R.; Ramón, D.J.; Yus, M. Easy α-alkylation of ketones with alcohols through a hydrogen autotransfer process catalyzed by RuCl2(dmso)4. Tetrahedron 2006, 62, 8988–9001.
- Martínez, R.; Ramón, D.J.; Yus, M. RuCl2(dmso)4 catalyzes the β-alkylation of secondary alcohols with primary alcohols through a hydrogen autotransfer process. Tetrahedron 2006, 62, 8982–8987.
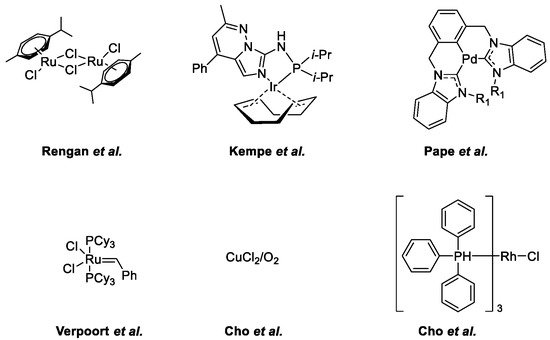
- Subramanian, M.; Sundar, S.; Rengan, R. Synthesis and structure of arene ruthenium(II) complexes: One-pot catalytic approach to synthesis of bioactive quinolines under mild conditions. Appl. Organomet. Chem. 2018, 32, e4582.
- Ruch, S.; Irrgang, T.; Kempe, R. New Iridium Catalysts for the Selective Alkylation of Amines by Alcohols under Mild Conditions and for the Synthesis of Quinolines by Acceptor-less Dehydrogenative Condensation. Chem. A Eur. J. 2014, 20, 13279–13285.
- Hahn, F.E.; Jahnke, M.C.; Pape, T. Synthesis of Pincer-Type Bis(benzimidazolin-2-ylidene) Palladium Complexes and Their Application in C−C Coupling Reactions. Organometallics 2007, 26, 150–154.
- Vander Mierde, H.; Van Der Voort, P.; De Vos, D.; Verpoort, F. A Ruthenium-Catalyzed Approach to the Friedländer Quinoline Synthesis. European J. Org. Chem. 2008, 2008, 1625–1631.
- Cho, C.S.; Ren, W.X.; Yoon, N.S. A recyclable copper catalysis in modified Friedländer quinoline synthesis. J. Mol. Catal. A Chem. 2009, 299, 117–120.
- Cho, C.S.; Seok, H.J.; Shim, S.O. A rhodium-catalyzed route for oxidative coupling and cyclization of 2-aminobenzyl alcohol with ketones leading to quinolines. J. Heterocycl. Chem. 2005, 42, 1219–1222.
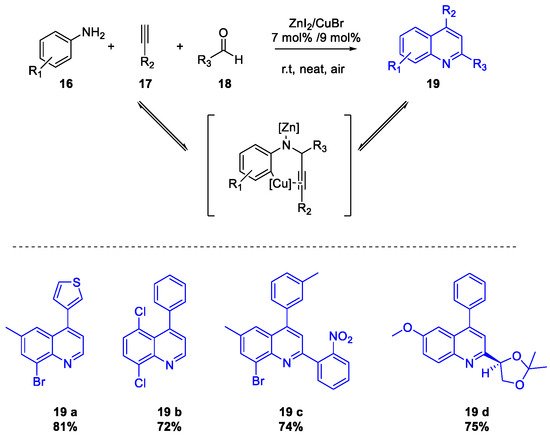
- Mondal, R.R.; Khamarui, S.; Maiti, D.K. CuBr–ZnI2 Combo-Catalysis for Mild Cu I –Cu III Switching and sp 2 C–H Activated Rapid Cyclization to Quinolines and Their Sugar-Based Chiral Analogues: A UV–Vis and XPS Study. ACS Omega 2016, 1, 251–263.
- Sarode, P.B.; Bahekar, S.P.; Chandak, H.S. Zn(OTf)2-mediated C H activation: An expeditious and solvent-free synthesis of aryl/alkyl substituted quinolines. Tetrahedron Lett. 2016, 57, 5753–5756.
- Korivi, R.P.; Cheng, C. Nickel-Catalyzed Cyclization of 2-Iodoanilines with Aroylalkynes: An Efficient Route for Quinoline Derivatives. J. Org. Chem. 2006, 71, 7079–7082.
- Cai, S.; Zeng, J.; Bai, Y.; Liu, X.-W. Access to Quinolines through Gold-Catalyzed Intermolecular Cycloaddition of 2-Aminoaryl Carbonyls and Internal Alkynes. J. Org. Chem. 2012, 77, 801–807.
- Zhu, M.; Fu, W.; Zou, G.; Xun, C.; Deng, D.; Ji, B. An efficient synthesis of 2-trifluoromethyl quinolines via gold-catalyzed cyclization of trifluoromethylated propargylamines. J. Fluor. Chem. 2012, 135, 195–199.
- Xu, X.; Su, H.; Bao, M.; Huang, J.; Qiu, L. Silver-Catalyzed Carbocyclization of Azide-Tethered Alkynes: Expeditious Synthesis of Polysubstituted Quinolines. Adv. Synth. Catal. 2018, 361, adsc.201801425.
- Wang, X.; Xie, P.; Sun, G.; Zhao, M.; Deng, Z.; Zhou, Y.; Bao, S. A systematic review and meta-analysis of the efficacy and safety of arbidol in the treatment of coronavirus disease 2019. Medicine (Baltimore) 2020, 99, e21402.
- Proskurnina, E.V.; Izmailov, D.Y.; Sozarukova, M.M.; Zhuravleva, T.A.; Leneva, I.A.; Poromov, A.A. Antioxidant potential of antiviral drug umifenovir. Molecules 2020, 25, 1577.
- Choudhary, S.; Silakari, O. Scaffold morphing of arbidol (umifenovir) in search of multi-targeting therapy halting the interaction of SARS-CoV-2 with ACE2 and other proteases involved in COVID-19. Virus Res. 2020, 289, 198146.
- Trofimov, F.A.; Tsyshkova, N.G.; Zotova, S.A.; Grinev, A.N. Synthesis of a new antiviral agent, arbidole. Pharm. Chem. J. 1993, 27, 75–76.
- Zhao, C.; Zhao, Y.; Chai, H.; Gong, P. Synthesis and in vitro anti-hepatitis B virus activities of some ethyl 5-hydroxy-1H-indole-3-carboxylates. Bioorg. Med. Chem. 2006, 14, 2552–2558.
- Cao, Z.; Dong, J. Preparation Method of Arbidol Hydrochloride. CN Patent CN 102351778A, February 2012.
- Mancuso, R.; Dalpozzo, R. Recent Progress in the Transition Metal Catalyzed Synthesis of Indoles. Catalysts 2018, 8, 458.
- Chaisan, N.; Kaewsri, W.; Thongsornkleeb, C.; Tummatorn, J.; Ruchirawat, S. PtCl 4 -catalyzed cyclization of N -acetyl-2-alkynylanilines: A mild and efficient synthesis of N -acetyl-2-substituted indoles. Tetrahedron Lett. 2018, 59, 675–680.
- Plosker, G.L. Telmisartan A Review of its Use in Hypertension. Drugs 2009, 69, 2477–2499.
- Yadav, G.; Ganguly, S. Structure activity relationship (SAR) study of benzimidazole scaffold for different biological activities: A mini-review. Eur. J. Med. Chem. 2015, 97, 419–443.
- Zhang, J.; Li, R.; Zhu, F.; Sun, C.; Shen, J. An improved synthesis of telmisartan via the copper-catalyzed cyclization of o -haloarylamidines. RSC Adv. 2020, 10, 13717–13721.
- Wang, P.; Zheng, G.; Wang, Y.; Wang, X.; Wei, H.; Xiang, W. Highly practical and cost-efficient synthesis of telmisartan: An antihypertensive drug. Tetrahedron 2012, 68, 2509–2512.
- Winkel-Shirley, B. Flavonoid Biosynthesis. A Colorful Model for Genetics, Biochemistry, Cell Biology, and Biotechnology. Plant Physiol. 2001, 126, 485–493.
- Zhao, X.; Zhou, J.; Lin, S.; Jin, X.; Liu, R. C–H Functionalization via Remote Hydride Elimination: Palladium Catalyzed Dehydrogenation of ortho-Acyl Phenols to Flavonoids. Org. Lett. 2017, 19, 976–979.
- Hui, D.S.; Azhar, E.I.; Madani, T.A.; Ntoumi, F.; Kock, R.; Dar, O.; Ippolito, G.; Mchugh, T.D.; Memish, Z.A.; Drosten, C.; et al. The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health—The latest 2019 novel coronavirus outbreak in Wuhan, China. Int. J. Infect. Dis. 2020, 91, 264–266.
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293.
- Nutho, B.; Mahalapbutr, P.; Hengphasatporn, K.; Pattaranggoon, N.C.; Simanon, N.; Shigeta, Y.; Hannongbua, S.; Rungrotmongkol, T. Why Are Lopinavir and Ritonavir Effective against the Newly Emerged Coronavirus 2019? Atomistic Insights into the Inhibitory Mechanisms. Biochemistry 2020, 59, 1769–1779.
- Liu, P.; Liu, H.; Sun, Q.; Liang, H.; Li, C.; Deng, X.; Liu, Y.; Lai, L. Potent inhibitors of SARS-CoV-2 3C-like protease derived from N-substituted isatin compounds. Eur. J. Med. Chem. 2020, 206, 112702.
- Horby, P.W.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Emberson, J.; Palfreeman, A.; Raw, J.; Elmahi, E.; Prudon, B.; et al. Lopinavir-ritonavir in patients admitted to hospital with COVID-19 (RECOVERY): A randomised, controlled, open-label, platform trial. Lancet 2020, 396, 1345–1352.
- Uzunova, K.; Filipova, E.; Pavlova, V.; Vekov, T. Insights into antiviral mechanisms of remdesivir, lopinavir/ritonavir and chloroquine/hydroxychloroquine affecting the new SARS-CoV-2. Biomed. Pharmacother. 2020, 131, 110668.
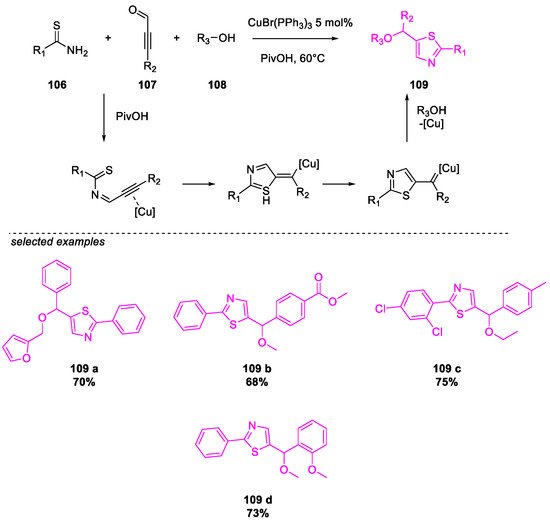
- Wang, Y.; Liu, X.; Zhu, B.; Guo, P.; Pei, Y.; He, Q.; Cao, H. Cu(I)-Catalyzed Three-Component Cyclization for the Construction of Functionalized Thiazoles. J. Org. Chem. 2020, 85, 10118–10124.
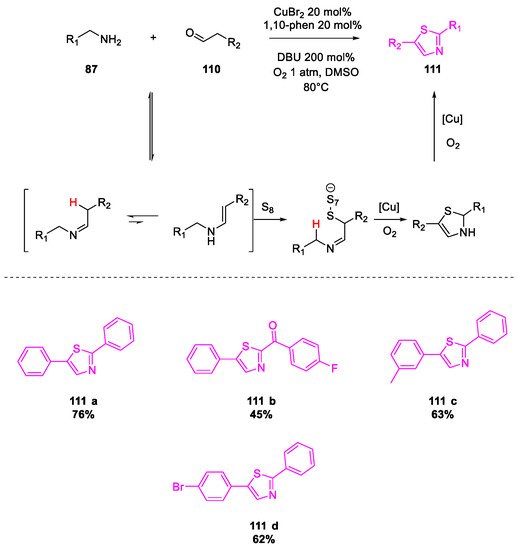
- Wang, X.; Qiu, X.; Wei, J.; Liu, J.; Song, S.; Wang, W.; Jiao, N. Cu-Catalyzed Aerobic Oxidative Sulfuration/Annulation Approach to Thiazoles via Multiple Csp 3–H Bond Cleavage. Org. Lett. 2018, 20, 2632–2636.
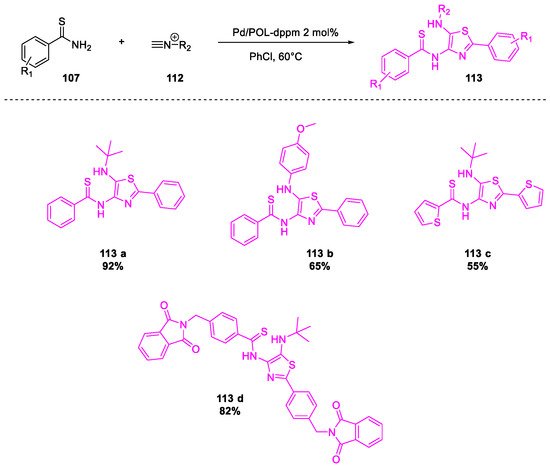
- Tong, W.; Li, W.-H.; He, Y.; Mo, Z.-Y.; Tang, H.-T.; Wang, H.-S.; Pan, Y.-M. Palladium-Metalated Porous Organic Polymers as Recyclable Catalysts for the Chemioselective Synthesis of Thiazoles from Thiobenzamides and Isonitriles. Org. Lett. 2018, 20, 2494–2498.
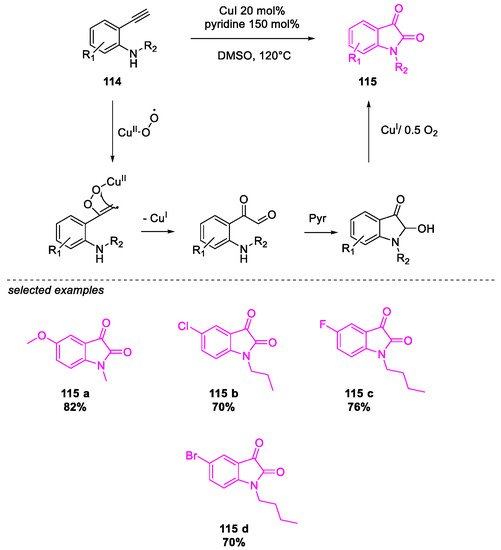
- Salvanna, N.; Ramesh, P.; Santosh Kumar, K.; Das, B. Copper-catalyzed aerobic oxidative intramolecular amidation of 2-aminophenylacetylenes: A domino process for the synthesis of isatin. New J. Chem. 2017, 41, 13754–13759.
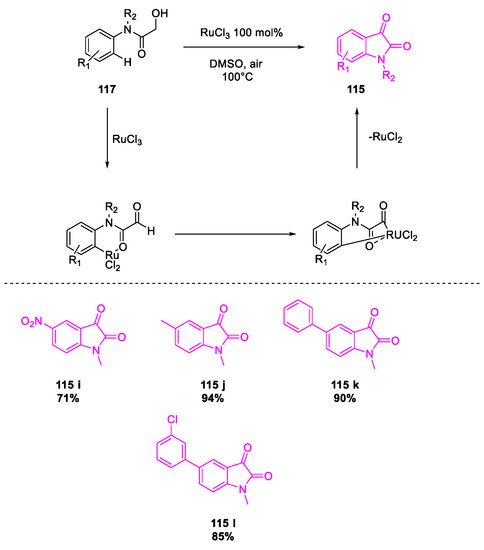
- Wang, Y.; Li, W.; Cheng, X.; Zhan, Z.; Ma, X.; Guo, L.; Jin, H.; Wu, Y. Ru(III)-mediated intramolecular ortho-C(sp2)–H activation/oxidative acylation: One-pot synthesis of isatins from α-hydroxy amides. Tetrahedron 2016, 72, 3193–3197.