Coupled signaling between bone-forming osteoblasts and bone-resorbing osteoclasts is crucial to the maintenance of bone homeostasis. We previously reported that v-crk avian sarcoma virus CT10 oncogene homolog-like (CrkL), which belongs to the Crk family of adaptors, inhibits bone morphogenetic protein 2 (BMP2)-mediated osteoblast differentiation, while enhancing receptor activator of nuclear factor kappa-B ligand (RANKL)-induced osteoclast differentiation. In this study, we investigated whether CrkL can also regulate the coupling signals between osteoblasts and osteoclasts, facilitating bone homeostasis. Osteoblastic CrkL strongly decreased RANKL expression through its inhibition of runt-related transcription factor 2 (Runx2) transcription. Reduction in RANKL expression by CrkL in osteoblasts resulted in the inhibition of not only osteoblast-dependent osteoclast differentiation but also osteoclast-dependent osteoblast differentiation, suggesting that CrkL participates in the coupling signals between osteoblasts and osteoclasts via its regulation of RANKL expression. Therefore, CrkL bifunctionally regulates osteoclast differentiation through both a direct and indirect mechanism while it inhibits osteoblast differentiation through its blockade of both BMP2 and RANKL reverse signaling pathways. Collectively, these data suggest that CrkL is involved in bone homeostasis, where it helps to regulate the complex interactions of the osteoblasts, osteoclasts, and their coupling signals.
1. Introduction
Bone is a complex and dynamic tissue, and it undergoes continuous renewal via bone remodeling processes to maintain appropriate bone mass and quality
[1]. These continuous processes of synthesis and destruction are fine-tuned by an equilibrium between bone-forming osteoblasts and bone-resorbing osteoclasts
[2][3][2,3]. In addition to the inherent functions of osteoblasts and osteoclasts, they contribute to each other’s functions via direct and indirect communication to maintain bone homeostasis
[4][5][6][7][4,5,6,7]. Moreover, osteoblasts can influence osteoclastic bone resorption by producing osteoclast regulatory factors, such as macrophage colony-stimulating factor (M-CSF), receptor activator of nuclear factor kappa-B ligand (RANKL), Fas ligand, complement component 3a, and semaphorins
[5][8][9][10][11][12][13][14][15][16][5,8,9,10,11,12,13,14,15,16]. Additionally, various osteoclast-derived factors, such as those released from the matrix, secreted from the osteoclast, and expressed on the cell membrane, can also influence the osteoblast differentiation and function
[16][17][16,17]. For example, matrix-derived factors, such as transforming growth factor β, BMP2, and insulin-like growth factors, which are released from osteoclastic bone resorption sites on the bone surface, stimulate the differentiation of the osteoblast progenitors. Osteoclasts also secrete products, such as BMP6, sphingosine-1-phosphate, and Wnt-10b to promote osteoblast precursor recruitment and differentiation. In addition, osteoclast membrane-bound factors, such as ephrin B2 and semaphorin D, are expected to support various interactions between the osteoclasts and mature osteoblasts promoting their osteoblastic activity
[17]. The communication between osteoclasts and osteoblasts is often the cause of the side effects of the presently available antiresorptive agents and anabolic agents for the treatment of bone disease
[18][19][20][21][22][18,19,20,21,22]. Therefore, a more detailed understanding of the cellular and molecular mechanisms of bone remodeling is necessary to identify and explore new therapeutic targets for bone disorders.
The cytoplasmic adaptor CrkL (v-crk avian sarcoma virus CT10 oncogene homolog-like) belongs to the Crk family of adaptors and is comprised of a single N-terminal Src homology 2 (SH2) domain and two consecutive SH3 domains (nSH3 and cSH3). CrkL is ubiquitously expressed in most tissues and exhibits several biological functions, such as adhesion, proliferation, migration, and survival
[23][24][25][26][23,24,25,26]. This adaptor protein can function via an interaction between its own SH2 or SH3 domain and numerous adaptor proteins, including paxillin, p130Cas, p120 c-cbl, insulin receptor substrate proteins, STAT5, and PI3K
[25][26][27][28][29][30][31][32][33][34][35][36][37][38][39][40][25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40]. We previously found that CrkII, another adaptor in the Crk family, plays a pivotal role in osteoclast and osteoblast differentiation
[41][42][41,42]. CrkII enhances osteoclast differentiation by activating Rac1, whereas it inhibits osteoblast differentiation via JNK activation
[41][42][41,42]. Furthermore, CrkII and CrkL exhibit overlapping functions in certain processes, including osteoclast and osteoblast differentiation, as they share several binding partners, due to remarkable homology between them. In contrast, several studies have reported that Crk proteins exhibit separate functions, notably during development
[43][44][43,44]. We previously reported that CrkL and CrkII show redundant function during osteoclast and osteoblast differentiation, as CrkL is a distinct gene transcribed from the CrkL locus but not Crk locus. The role of CrkL in communication between osteoclasts and osteoblasts during bone remodeling processes remains unknown; therefore, in the present study, we thoroughly investigated the role of CrkL during bone remodeling by considering various aspects.
2.Bifunctional Role of CrkL during Bone Remodeling
2.1. CrkL Has a Positive and Negative Effect on Osteoclast and Osteoblast Differentiation, Respectively
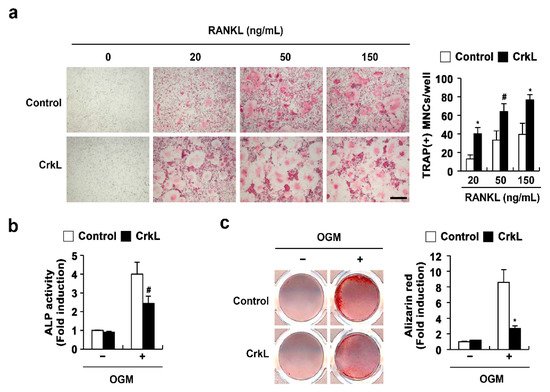
We previously reported that CrkL and CrkII exhibit overlapping functions in osteoclasts and osteoblasts. In the present study, to confirm the roles of CrkL in osteoclasts and osteoblasts, we determined the effects of retrovirus-mediated overexpression of CrkL in bone marrow-derived monocyte/macrophage lineage cells (BMMs) and primary osteoblasts. The formation of large multinucleated osteoclasts, induced by RANKL, was significantly enhanced in BMMs overexpressing CrkL compared to that in control (
Figure 1a); however, alkaline phosphate (ALP) activity and bone mineralization induced by osteogenic media (OGM) was significantly inhibited in osteoblasts overexpressing CrkL (
Figure 1b). These results confirmed that CrkL upregulates RANKL-mediated osteoclast differentiation, while it downregulates osteoblast differentiation and function.
Figure 1. CrkL enhances osteoclast differentiation, while it inhibits osteoblast differentiation. (a) TRAP staining images. Osteoclast differentiation of BMMs overexpressing the control or CrkL retrovirus following treatment with M-CSF, or M-CSF and RANKL (left panel). Number of TRAP-positive multinuclear cells (right panel). Bar: 200 µm. (b,c) Primary osteoblast precursors overexpressing the control or CrkL retrovirus were cultured in OGM. (b) ALP activity assay. (c) Alizarin red staining images (left panel). Quantification of alizarin red staining intensities (right panel). # p < 0.05; * p < 0.01 as compared with the controls.
2.Bifunctional Role of CrkL during Bone Remodeling
2.2. CrkL Indirectly Inhibits Osteoclast Differentiation by Regulating RANKL (Tnfsf11) Expression
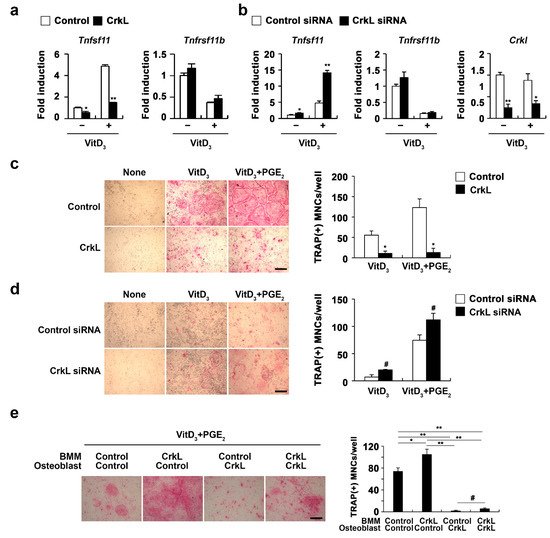
Osteoblasts produce RANKL and osteoprotegerin (OPG), that contribute to bone homeostasis via the regulation of osteoclastogenesis
[15]. We further examined whether CrkL is associated with osteoblast-mediated osteoclast differentiation. Overexpression of CrkL in osteoblasts significantly inhibited the mRNA expression of
Tnfsf11 in both nonstimulated and 1,25 (OH)
2 vitamin D
3 (Vit D
3)-stimulated osteoblasts without affecting the OPG (
Tnfrsf11b) expression (
Figure 2a). Conversely, downregulation of CrkL by siRNA led to an increase in the mRNA levels of
Tnfsf11 (
Figure 2b). To functionally validate the role of CrkL in osteoblastic RANKL expression, we cocultured the osteoblasts with osteoclast precursor cells. The coculture of osteoblasts overexpressing or downregulating CrkL with osteoclast precursor cells displayed decreased or increased osteoclast formation, in contrast to each control osteoblast culture (
Figure 2c,d).
Figure 2. CrkL attenuates RANKL expression. (a) Primary osteoblast precursors overexpressing the control or CrkL retrovirus in the presence or absence of Vit D3. mRNA expression of the indicated genes. (b) Primary osteoblast precursors were transfected with control or CrkL siRNAs in the presence or absence of Vit D3. mRNA expression of the indicated genes was assessed by real-time PCR. (c) Primary osteoblast precursors overexpressing the control or CrkL retrovirus were cocultured with BMMs and Vit D3 in the presence or absence of PGE2. TRAP staining images (left panel) where used to enumerate the number of TRAP-positive multinuclear cells (right panel). Bar: 200 µm. (d) Primary osteoblast precursors transfected with control or CrkL siRNAs and cocultured with BMMs and Vit D3 in the presence or absence of PGE2. TRAP staining images (left panel) where used to enumerate the number of TRAP-positive multinuclear cells (right panel). Bar: 200 µm. (e) Osteoclast differentiation in BMMs, cocultured with osteoblasts, treated as indicated in the presence of Vit D3 and PGE2. TRAP staining images (left panel) where used to enumerate the number of TRAP-positive multinuclear cells (right panel) in each sample. Bar: 200 µm. # p < 0.05; * p < 0.01; ** p < 0.001 as compared with the controls.
To further assess the role of CrkL in osteoblast-mediated osteoclast differentiation, osteoblast and osteoclast precursor cells were transduced with control or CrkL retrovirus, as illustrated in
Figure 2e, and then cocultured in the presence of vitamin D
3 (Vit D
3) and prostaglandin E
2 (PGE
2). Interestingly, a significant decrease in osteoclast formation was observed when BMMs overexpressing CrkL were cocultured with osteoblasts overexpressing CrkL, compared to the control (
Figure 2e). These results indicated that CrkL inhibits osteoblast-mediated osteoclast differentiation by blocking RANKL expression, though it increases RANKL-mediated osteoclast differentiation in osteoclast precursor cells.
2.3. CrkL Inhibits RANKL-Mediated Osteoblast Differentiation
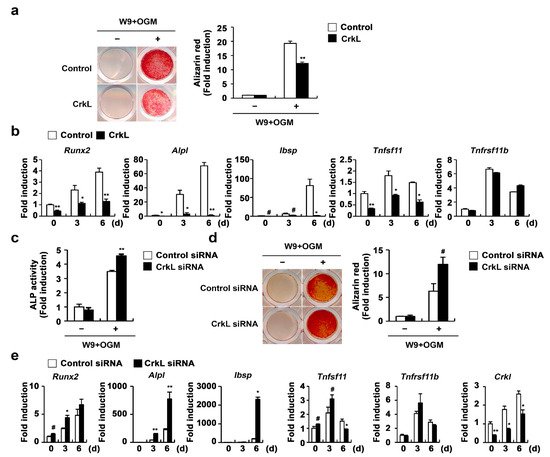
Recently, it has been reported that vesicular receptor activator of nuclear factor kappa-B (RANK) secreted from mature osteoclasts activates RANKL reverse signaling in osteoblasts and enhances osteoblast differentiation
[45]. Moreover, the W9 peptide, which is known to bind RANKL and inhibits RANKL-induced osteoclast differentiation in vitro, also binds RANKL on osteoblasts and promotes osteoblast differentiation, presumably via RANKL reverse signaling
[46][47][48][46,47,48]. Therefore, we tested the effects of CrkL on W9-induced osteoblast differentiation, to evaluate whether the inhibition of RANKL expression in osteoblasts caused by CrkL affects osteoblast differentiation by regulating RANKL reverse signaling. As illustrated in
Figure 3, overexpression of CrkL in osteoblasts significantly inhibited W9-induced bone mineralization. Consistent with bone mineralization, expression of typical osteogenic marker genes, including Runx2, alkaline phosphatase (
Alpl), and bone sialoprotein (
Ibsp), was significantly inhibited by CrkL overexpression (
Figure 3a,b). In contrast, downregulation of endogenous CrkL expression markedly increased the ALP activity, bone mineralization, and expression of typical osteogenic marker genes (
Figure 3c–e). Collectively, these results indicate that osteoblastic CrkL regulates osteoblast differentiation by inhibiting both BMP2 signaling and RANKL reverse signaling.
Figure 3. CrkL inhibits RANKL–RANK reverse signaling pathway. (a,b) Primary osteoblast precursors overexpressing the control or CrkL retrovirus were cultured in OGM supplemented with W9. (a) Alizarin red staining images (left panel) allowed for the quantification of the alizarin red staining intensities (right panel) under each condition. (b) mRNA expression of the indicated genes. (c–e) Primary osteoblast precursors transfected with control or CrkL siRNAs were cultured in OGM with W9. (c) ALP activity assay. (d) Alizarin red staining images (left panel). Quantification of alizarin red staining intensities (right panel). (e) mRNA expression of the indicated genes. # p < 0.05; * p < 0.01; ** p < 0.001 as compared with the controls.
2.4. CrkL Inhibits RANKL Expression via Interaction with Runx2
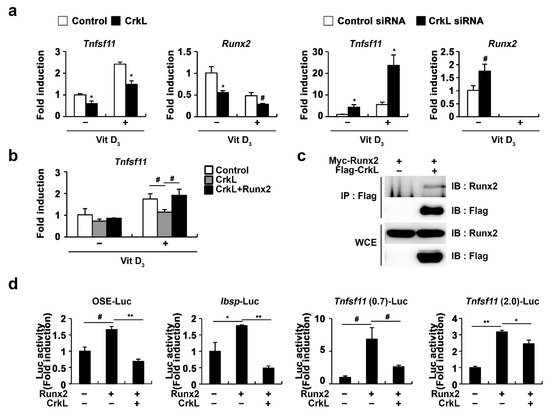
As Runx2 has been implicated in RANKL expression in various cell types, such as prostate cancer cells, vascular smooth muscle cells, and osteoblasts
[49][50][51][52][49,50,51,52], we further examined whether CrkL-inhibited
Tnfsf11 expression is involved in the regulation of Runx2 expression. As illustrated in
Figure 4a, Runx2 expression controlled by CrkL was similar to that of
Tnfsf11. Moreover, the expression of
Tnfsf11 as well as that of Runx2 was suppressed when CrkL was overexpressed, whereas it was increased when CrkL was downregulated. Moreover, inhibition of
Tnfsf11 expression by CrkL was rescued by the forced expression of Runx2 (
Figure 4b). In order to gain a deeper insight into the mechanisms underlying the inhibition of
Tnfsf11 expression by CrkL, we examined whether CrkL directly interacts with Runx2. Coimmunoprecipitation revealed the direct interaction between CrkL and Runx2 in HEK-293T cells (
Figure 4c). Furthermore, Runx2 induced the expression of 6XOSE (Runx2 DNA-binding elements),
Ibsp, and
Tnfsf11 promoter reporter, and its effects were significantly inhibited by CrkL (
Figure 4d). Collectively, these results indicated that the CrkL-induced decreased expression of RANKL is mediated via the suppression of Runx2 transcriptional activity.
Figure 4. CrkL inhibits Runx2 transcriptional activity. (a) Primary osteoblast precursors overexpressing control or CrkL retrovirus and primary osteoblast precursors transfected with control or CrkL-siRNAs were cultured in the presence or absence of Vit D3. (b) Primary osteoblast precursors overexpressing control, CrkL or CrkL and Runx2 were cultured in the presence or absence of Vit D3. mRNA expression of Tnfsf11. (c) Co-immunoprecipitation assays in Runx2 or Runx2 and CrkL-transfected HEK-293T cells. (d) Luciferase assay of HEK-293T cells transfected with various expression plasmids. # p < 0.05; * p < 0.01; ** p < 0.001 as compared with the controls.
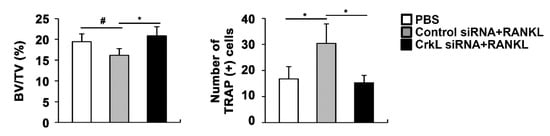
2.5. Downregulation of CrkL Can Protect RANKL-Induced Bone Loss In Vivo
Eventually, we assessed the local administration of siRNA, targeting CrkL, in a mouse calvaria model. In microcomputed tomography (µCT) analyses, the injection of RANKL to the calvaria led to a significant decrease in bone mass. RANKL-induced bone loss was attenuated by the local administration of CrkL siRNA (
Figure 5). These results indicated that CrkL may be a potential target in the development of new therapeutics for bone diseases.
Figure 5. CrkL knockdown prevents RANKL-induced bone loss. Bone volume to tissue volume (BV/TV) and the number of TRAP-positive cells in the murine calvarial model treated with PBS, RANKL and control, or CrkL siRNA. # p < 0.05; * p < 0.01 as compared with the controls.
3. Conclusions
CrkL inhibits BMP2-mediated osteoblast differentiation of the osteoblast precursors, while enhancing RANKL-induced osteoclast differentiation of the osteoclast precursors. CrkL also affects both osteoblast-dependent osteoclast differentiation and osteoclast-dependent osteoblast differentiation via its suppression of RANKL expression in osteoblasts. Further evaluations of the in vivo roles of CrkL during bone remodeling will help to understand the potential therapeutic value of CrkL in the future treatment of various bone diseases.