Gut microbiota derived metabolites can act as a mediator of microbial influence through circulation on various target organs, including the cardiovascular systems. Thus far, scientists have proposed several mechanisms by which dysbiotic gut microbiota contributes to CVD, such as alterations of microbiota-derived metabolite short-chain fatty acids (SCFA), increases of trimethylamine-N-oxide (TMAO), inhibition of nitric oxide (NO), and aberrant activation of the renin-angiotensin system (RAS). A meta-analysis study summarized 19 studies with 19,256 participants and found that individuals with high concentrations of TMAO and its precursors were associated with increased risks of major adverse cardiovascular events and all-cause mortality.
- aryl hydrocarbon receptor
- cardiovascular disease
- hypertension
- gut microbiota
- postbiotics
- short chain fatty acid
- prebiotics
- probiotics
- trimethylamine-N-oxide
- developmental origins of health and disease (DOHaD)
1. Introduction
Cardiovascular diseases (CVDs), a cluster of disorders of the heart and blood vessels, are the leading cause of death worldwide [1]. The developmental origins of health and disease (DOHaD) theory posits that exposure to various insults during critical periods in fetal development leads to structural changes and functional adaption, resulting in increased risk of adult diseases, including CVDs [3][2]. Important support for the DOHaD concept came from epidemiological reports following birth cohorts in severe famines, which demonstrated that malnutrition during gestation induced a cluster of CVD risk factors, such as hypertension, dyslipidemia, obesity, kidney disease, type 2 diabetes, and cardiovascular morbidity in adult offspring [4,5][3][4]. –child dyads reported that a better gestational CV health score in the mothers was associated with better CV health in the offspring from ages 10 to 14 years [6][5].
Emerging evidence supports that gut microbiota may indirectly or directly influence cardiovascular risk [7,8,9][6][7][8]. A meta-analysis study summarized 19 studies with 19,256 participants and found that individuals with high concentrations of TMAO and its precursors were associated with increased risks of major adverse cardiovascular events and all-cause mortality [12][9]. Maternal nutritional insults have been shown to alter gut microbiota balance, resulting in an increased risk of adult diseases [13][10]. However, relatively little is known about whether (and how) various early life insults could affect gut microbiota, resulting in CVDs in adult offspring.
Nevertheless, developmental programming, besides determining the risk for CVDs in adulthood, also offers an innovative approach to prevent CVDs by so-called reprogramming [14][11]. By switching therapy from adulthood to early life before disease occurs, we have the potential to postpone or reduce undesirable programming processes that would lead to CVDs. Over the year, the gut microbiota has gained more attention because, unlike non-modifiable CV risk factors, it can be modified through agents that modulate the intestinal bacterial flora, including prebiotics, probiotics, synbiotics, etc. Accordingly, one may assume that early gut microbiota-targeted therapy may serve as a reprogramming strategy to prevent the developmental origins of CVD.
The purpose of our scoping review is to provide insight on gut microbiota implicated in the developmental programming of CVD. Hence, we examine mechanisms linking gut microbiota to cardiovascular programming that, in light of the available evidence, can be considered therapeutic targets. In particular, we focus on addressing probiotics, prebiotics, and postbiotics as a reprogramming strategy for prevention of developmental programming of CVD.
We searched the PubMed/MEDLINE databases for studies published in English between January 1980 and May 2021, using the following search terms: “cardiovascular disease”, “developmental programming”, “DOHaD”, “reprogramming”, “gut microbiota”, “probiotics”, “prebiotics”, “synbiotics”, “postbiotics”, “mother”, “pregnancy”, “gestation”, “offspring”, “progeny”, “atherosclerosis”, “heart”, “vascular”, “endothelial dysfunction”, “stroke”, “thrombosis”, “aryl hydrocarbon receptor” and “hypertension”. Additional studies were then selected and assessed based on fitting references in eligible papers. However, only approximately 10% belong to the research of DOHaD. Among them, hypertension accounts for nearly 80% of searchable publications.
2. Developmental Programming of CVD: Human Evidence
Additionally, extensive evidence indicates that other early life adverse influences contribute to CVD in later life, including maternal illness, pregnancy complications, medication use in pregnancy, and in utero exposure to environmental pollutants [20,21,22][12][13][14]. Prior research also suggested an association between medication use in pregnant women, such as glucocorticoid [24][15] and non-steroidal anti-inflammatory drugs [25][16], and adverse cardiovascular renal outcomes in their progeny. Several other risk factors in early life affecting cardiovascular outcomes in offspring have been reported, such as vitamin D deficiency [26][17], gestational hypertension [27][18], short-term breastfeeding [28][19], and excessively rapid weight gain postnatally [29][20]. Moreover, early life toxic environmental exposure, such as endocrine-disrupting chemicals, is linked to cardiometabolic traits in childhood [30][21].
However, epidemiological studies often do not provide information in which a direct cause–effect relationship can be established. Instead, the results from animal models can help one understand which developmental stage is critical for cardiovascular programming, to discover molecular mechanisms underlying the developmental origins of CVD, and to develop novel reprogramming strategies.
3. Implications of Gut Microbiota in the Developmental Origins of CVD
Animal models have provided compelling evidence that adverse early life conditions induce cardiovascular programming, coinciding with altered gut microbiota. Table 1 summarizes animal studies documenting the association between gut microbiota, early life insults, and subsequent CVDs in adult offspring [34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51][22][23][24][25][26][27][28][29][30][31][32][33][34][35][36][37][38][39]. CVDs often occur after a prolonged asymptomatic phase in childhood. For the sake of brevity, the present review is solely restricted to data obtained from adult offspring.
| Animal Models | Cardiovascular Outcomes | Programming Mechanisms Related Gut Microbiota | Species/ Gender |
Age at Measure | Ref. |
|---|---|---|---|---|---|
| Maternal high-fructose diet | Hypertension | Decreased SCFA receptor GPR41 and GPR43 expression | SD rat/M | 12 weeks | [22] |
| Maternal high-fructose diet | Hypertension | Decreased plasma TMA level; reduced phylum Verrucomicrobia and genus Akkermansia abundance | SD rat/M | 12 weeks | [23] |
| Maternal plus post-weaning high-fructose diet | Hypertension | Decreased abundance of genera Bacteroides, Dysgonomonas, and Turicibacter | SD rat/M | 12 weeks | [24] |
| Maternal high-fructose diet and TCDD exposure | Hypertension | Increased abundance of genus Gordonibacter | SD rat/M | 12 weeks | [25] |
| Maternal adenine-induced chronic kidney disease | Hypertension | A decreased α-diversity and an increased F/B ratio; A decreased abundance of the genus Bifidobacterium | SD rat/M | 12 weeks | [26] |
| Maternal minocycline administration | Hypertension | An increase F/B ratio, and decreased genera Lactobacillus, Ruminococcus, and Odoribacter abundance | SD rat/M | 12 weeks | [27] |
| Maternal ADMA and TMAO exposure | Hypertension | Decreased abundance of family Erysipelotrichaceae | [28] | ||
| Maternal hypertension | Hypertension | An increased abundance of the genera Bifidobacterium, Lactobacillus, Turicibacter, and Akkermansia | SHR/M | 12 weeks | [29] |
| Maternal hypertension | Hypertension | An increased F/B ratio | SHR/M | 12 weeks | [30] |
| Maternal high-fat diet | Obesity and insulin resistance | Decreased gut microbiota richness | C57BL/6J mouse/M and F | 12 weeks | [31] |
| Maternal high-fat diet | Obesity and nonalcoholic fatty liver disease | Decreased α-diversity | C57BL/6J mouse/M and F | 17 weeks | [32] |
| Maternal high-fat and high-cholesterol diet | Hypertension, endothelial dysfunction, increased lipid profile and insulin resistance | Decreased α-diversity | Wistar rat/M | 90 days | [33] |
| Maternal plus post-weaning high-fat diet | Hypertension | An increased F/B ratio; a reduction of genera Lactobacillus and Akkermansia | SD rat/M | 16 weeks | [34][35] |
| Maternal L-NAME administration plus post-weaning high-fat diet | Hypertension | An increased F/B ratio | SD rat/M | 16 weeks | [36] |
| Maternal Western-style diet | Obesity and nonalcoholic fatty liver disease | An increase in abundance of genus Ruminococcus | C57BL/6J mouse/M | 20 weeks | [37] |
| Maternal dyslipidemia | Hypertension and increased lipid profile | A decrease of genera Lactobacillus | Wistar rat/M and F | 24 weeks | [38] |
| Prenatal androgen exposure | Hypertension, decreased heart rate, obesity, and increased thickness of left ventricle | An increased abundance of bacteria associated with production of SCFAs. | Wistar rat/F | 4 months | [39] |
A variety of environmental insults in early life were reported to induce cardiovascular programming related to alterations of gut microbiota, including maternal high-fructose diet [34,35[22][23][24],36], maternal high-fructose diet plus 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) exposure [37][25], maternal chronic kidney disease [38][26], maternal minocycline administration [39][27], maternal asymmetric dimethylarginine (ADMA), trimethylamine N-oxide (TMAO) exposure [40][28], maternal hypertension [41[29][30],42], maternal high-fat and/or post-weaning high-fat diet [43[31][32][33][34][35],44,45,46,47], maternal NG-nitro-L-arginine-methyl ester (L-NAME) administration plus post-weaning high-fat diet [48][36], maternal Western-style diet [49][37], maternal dyslipidemia [50][38], and prenatal androgen exposure [51][39].
The most common cardiovascular outcome being studied in altered gut microbiota-related animal models is hypertension [34[22][23][24][25][26][27][28][29][30][33][34][35][36][38][39],35,36,37,38,39,40,41,42,45,46,47,48,50,51], followed by endothelial dysfunction [44][32] and ventricular hypertrophy [51][39]. Abnormalities in gut microbiota were also related CV risks, such as obesity [43,44[31][32][37][39],49,51], insulin resistance [43[31][33],45], increased lipid profile [45[33][38],50], and nonalcoholic fatty liver disease [44,48][32][36]. Abnormalities in gut microbial richness and diversity have been linked to a higher risk of developing other CVDs, such as coronary artery disease, cardiomyopathy, and heart failure [52][40]. However, little information exists regarding the role of gut microbiota in the developmental origins of those CVDs.
AsTable 1illustrates, the rat is the most common species used. The possible reason might be that rat models provide a low-cost option with a short life cycle that is easy to handle [53,54][41][42]. Although other species, such as rabbits, sheep, and pigs, are used in DOHaD research [53[41][42],54], limited information is provided on using large animals to study gut microbiota and the developmental origins of CVD.
In adulthood, every one month of the rat is equivalent to three human years [ the timing of cardiovascular outcomes determined from 12 weeks to 4 months of age in rats, which corresponds to humans from childhood to early adulthood. Accordingly, knowledge gaps about long-term adverse effects of early life insults on CVD and gut microbiota in older adult offspring remain large.
In view of the fact that a variety of insults during fetal development generates similar cardiovascular outcomes (e.g., hypertension) in adulthood, this raises the possibility that a common pathway is involved in the pathogenesis of the developmental origins of CVD. Presently, several mechanisms have been linked to the developmental origins of CVD, such as oxidative stress, nitric oxide (NO) deficiency, Some of them are interrelated to gut microbiota dysbiosis. Recent evidence has been accumulated by deciphering the role of gut microbiota in the developmental origins of CVD, including alterations of SCFAs and their receptors, increases of TMAO, uremic toxins, and aryl hydrocarbon receptor (AhR), and aberrant renin-angiotensin system (RAS) [10,11][43][44] (Figure 1).
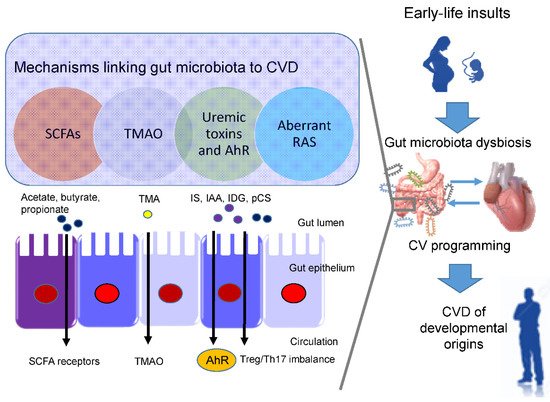
Short chain fatty acids (SCFAs) are formed during bacterial fermentation of carbohydrates in the gut. SCFAs have one to six carbon atoms (C1-C6), mainly consisting of acetate (C2), propionate (C3), and butyrate (C4) [56][45]. SCFAs are generally known to induce vasodilatation, in favor of antihypertension [56][45].
In spontaneously hypertensive rats (SHRs), hypertension is related to decreased abundance of acetate- and butyrate-producing bacteria [57][46]. SCFAs exert their regulation on BP mainly by activating their SCFA receptors, including G protein-coupled receptor 41 (GPR41), 43 (GPR43), 109A (GRP109A), and olfactory receptor 78 (Olfr78) [56][45]. The Olfr78 is expressed in the renal juxtaglomerular apparatus, where it mediates renin secretion in response to SCFAs to elevate BP [58][47]. In addition to hypertension, the metabolism of SCFAs is also a factor contributing to obesity [59][48].
In a maternal minocycline exposure model, minocycline-induced hypertension is associated with decreased plasma levels of acetate and butyrate [39][27]. Another report demonstrated that maternal garlic oil therapy protects against increases in blood pressure induced by high-fat diets in adult male rat offspring, which coincided with increases of acetate, butyrate, and propionate levels, as well as their producing microorganisms [60][49]. Additionally, SCFA supplementation in pregnancy and lactation have shown positive benefits on hypertension of developmental origins [35,61][23][50]. These findings support the notion that SCFAs and their receptors might be a key mechanism underlying developmental programming of hypertension.
Besides CVD, TMAO also contributes to other cardiovascular risks, such as chronic kidney disease (CKD), type II diabetes, insulin resistance, and NAFAD [63][51]. ) signaling, inflammatory gene expression, and leukocyte-endothelial cell adhesion in atherosclerosis development [64][52]. TMAO is a small colorless amine oxide produced by gut microbiota metabolism. The first step involves gut microbial trimethylamine (TMA) formation from dietary precursors (e.g., choline and carnitine); in the second step, TMA is oxidized to TMAO by flavin-containing monooxygenases (FMOs) in the liver
Maternal TMAO administration can induce hypertension in adult male offspring [40][28]. Conversely, 3,3-dimethyl-1-butanol (DMB, a TMA inhibitor) treatment during pregnancy and lactation protected adult offspring against hypertension programmed by a maternal high-fructose diet, which was associated with the reduction of TMA and TMAO levels [35][23]. Moreover, perinatal resveratrol therapy prevented maternal CKD-induced hypertension in adult male rat offspring and was associated with a decreased TMAO-to-TMA ratio [38][26]. These observations suggest a pathogenic association between the TMA-TMAO pathway and the developmental origins of CVD.
One example is TMAO, a cardiovascular risk factor and uremic toxins. Additionally, several tryptophan metabolites are known as gut microbiota-derived uremic toxins, such as indoxyl sulfate Among them, IS and IAA were related to the risk for cardiovascular morbidity and mortality in uremic patients [69][53]. In CKD, IS can activate monocytes, intensify inflammatory process, augment oxidative stress and impair hemostatic system, all of which are major contributors to the development of CVD [70,71][54][55].
Moreover, IAA and several microbial tryptophan catabolites are ligands for aryl AhR [72][56]. (TH17) axis and triggers inflammation, by which activation of AhR by its ligands is closely associated with the development of CVD [73,74][57][58]. An imbalance of T regulatory cells (Treg) and TH17 cells is involved in the development of hypertension [75][59]. In patients with CKD, Treg/Th17 imbalance has been associated with the pathogenesis of cardiovascular complications [76][60].
In a maternal CKD-induced hypertension model, maternal tryptophan therapy preventing offspring hypertension was associated with mediation of the AhR signaling pathway [77][61]. On the other hand, antagonizing AhR signaling by resveratrol has been reported to protect adult offspring against hypertension in several hypertension models of developmental origins [36,78,79][24][62][63]. Prior research demonstrated that IS and AhR both can promote thrombosis [80,81][64][65]. As thrombus formation, secondary to atherosclerotic plaque disruption, plays a major role in the development of several CVDs [82][66], attention will need to be paid to better understand the interplay between AhR and uremic toxins underlying developmental programming of thrombotic-related CVDs.
Another uremic toxin is p-Cresyl sulfate (pCS), derived from aromatic amino acids metabolized by gut bacteria [83][67]. Increased levels of pCS have been related to worsening CV outcomes in patients with CKD [83][67]. Previous work demonstrated that p-Cresyl sulfate could increase expression of proinflammatory cytokines and adhesion molecules, therefore mechanistically promoting atherogenesis [84][68].
Within the RAS, regulation is achieved through a cascade of proteases that generate several bioactive peptides [86][69]. Activation of the classical RAS triggers vasoconstriction and inflammation under pathophysiological conditions, thus promoting hypertension and cardiovascular damage [85][70]. Emerging evidence suggests that aberrant RAS plays a key role in cardiovascular programming and RAS-based interventions can be used as a reprogramming strategy to prevent DOHaD-related disorders [86][69]. Treating young offspring with renin inhibitor aliskiren [87][71], ACE inhibitor captopril [88][72], or angiotensin receptor blocker (ARB) losartan [89][73] between 2 and 4 weeks of age was shown to protect against hypertension programmed by various maternal insults in adulthood.
Angiotensin-converting enzyme 2 (ACE2), a homologue of ACE, converts ANG II to ANG-(1–7) Via regulation of intestinal amino acid transport, previous research showed that ACE2 plays a crucial non-catalytic role in gut biology and modulation of gut microbiota [91][74]. One previous study reported that administration with the ACE2 activator or with ANG-(1–7) during pregnancy could alleviate cardiovascular dysfunction in adult SHR offspring [92][75]. Another report showed that the antihypertensive effect of probiotics might be due to their ability to produce ACE inhibitory peptides [93][76].
Since gut microbiome dysbiosis has been connected to CVD by modulating the gut RAS [94][77], these findings support the notion that the interplay between gut microbiota and the RAS implicates the pathogenesis of cardiovascular programming, although this remains speculative.
References
- World Health Organization. The Top 10 Causes of Death. 2020. Available online: (accessed on 9 December 2020).
- Hanson, M.; Gluckman, P. Developmental origins of noncommunicable disease: Population and public health implications. Am. J. Clin. Nutr. 2011, 94, 1754S–1758S.
- Roseboom, T.; de Rooij, S.; Painter, R. The Dutch famine and its long-term consequences for adult health. Early Hum. Dev. 2006, 82, 485–491.
- Luyckx, V.A.; Bertram, J.F.; Brenner, B.M.; Fall, C.; Hoy, W.E.; Ozanne, S.E.; Vikse, B.E. Effect of fetal and child health on kidney development and long-term risk of hypertension and kidney disease. Lancet 2013, 382, 273–283.
- Perak, A.M.; Lancki, N.; Kuang, A.; Labarthe, D.R.; Allen, N.B.; Shah, S.H.; Lowe, L.P.; Grobman, W.A.; Lawrence, J.M.; Lloyd-Jones, D.M.; et al. HAPO Follow-up study cooperative research group. Associations of maternal cardiovascular health in pregnancy with offspring cardiovascular health in early adolescence. JAMA 2021, 325, 658–668.
- Scarmozzino, F.; Poli, A.; Visioli, F. Microbiota and cardiovascular disease risk: A scoping review. Pharmacol. Res. 2020, 159, 104952.
- Tang, W.W.; Kitai, T.; Hazen, S.L. Gut microbiota in cardiovascular health and disease. Circ. Res. 2017, 120, 1183–1196.
- Tang, W.H.W.; Bäckhed, F.; Landmesser, U.; Hazen, S.L. Intestinal microbiota in cardiovascular health and disease: JACC State-of-the-Art review. J. Am. Coll. Cardiol. 2019, 73, 2089–2105.
- Heianza, Y.; Ma, W.; Manson, J.E.; Rexrode, K.M.; Qi, L. Gut microbiota metabolites and risk of major adverse cardiovascular disease events and death: A systematic review and meta-analysis of prospective studies. J. Am. Heart Assoc. 2017, 6, e004947.
- Chu, D.M.; Meyer, K.M.; Prince, A.L.; Aagaard, K.M. Impact of maternal nutrition in pregnancy and lactation on offspring gut microbial composition and function. Gut Microbes 2016, 7, 459–470.
- Tain, Y.L.; Joles, J.A. Reprogramming: A preventive strategy in hypertension focusing on the kidney. Int. J. Mol. Sci. 2015, 17, 23.
- Thornburg, K.L. The programming of cardiovascular disease. J. Dev. Orig. Health Dis. 2015, 6, 366–376.
- Santos, M.S.; Joles, J.A. Early determinants of cardiovascular disease. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 581–597.
- Tain, Y.L.; Hsu, C.N. Interplay between oxidative stress and nutrient sensing signaling in the developmental origins of cardiovascular disease. Int. J. Mol. Sci. 2017, 18, 841.
- Dalziel, S.R.; Walker, N.K.; Parag, V.; Mantell, C.; Rea, H.H.; Rodgers, A.; Harding, J.E. Cardiovascular risk factors after antenatal exposure to betamethasone: 30-year follow-up of a randomised controlled trial. Lancet 2005, 365, 1856–1862.
- Antonucci, R.; Zaffanello, M.; Puxeddu, E.; Porcella, A.; Cuzzolin, L.; Pilloni, M.D.; Fanos, V. Use of non-steroidal anti-inflammatory drugs in pregnancy: Impact on the fetus and newborn. Curr. Drug Metab. 2012, 13, 474–490.
- Hrudey, E.J.; Reynolds, R.M.; Oostvogels, A.J.J.M.; Brouwerv, I.A.; Vrijkotte, T. The association between maternal 25-hydroxyvitamin D concentration during gestation and early childhood cardio-metabolic outcomes: Is there interaction with pre-pregnancy bmi? PLoS ONE 2015, 10, e0133313.
- Fraser, A.; Nelson, S.M.; Macdonald-Wallis, C.; Sattar, N.; Lawlor, D. Hypertensive disorders of pregnancy and cardiometabolic health in adolescent offspring. Hypertension 2013, 62, 614–620.
- Hosaka, M.; Asayama, K.; Staessen, J.A.; Ohkubo, T.; Hayashi, K.; Tatsuta, N.; Kurokawa, N.; Satoh, M.; Hashimoto, T.; Hirose, T.; et al. Breastfeeding leads to lower blood pressure in 7-year-old Japanese children: Tohoku study of child development. Hypertens. Res. 2012, 36, 117–122.
- Keijzer-Veen, M.G.; Finken, M.J.J.; Nauta, J.; Dekker, F.W.; Hille, E.T.; Frölich, M.; Wit, J.M.; Van Der Heijden, A. Is blood pressure increased 19 years after intrauterine growth restriction and preterm birth? A prospective follow-up study in the Netherlands. Pediatrics 2005, 116, 725–731.
- Tang-Peronard, J.L.; Andersen, H.R.; Jensen, T.K.; Heitmann, B.L. Endocrine-disrupting chemicals and obesity development in humans: A review. Obes. Rev. 2011, 12, 622–636.
- Hsu, C.-N.; Lin, Y.-J.; Hou, C.-Y.; Tain, Y.-L. Maternal administration of probiotic or prebiotic prevents male adult rat offspring against developmental programming of hypertension induced by high fructose consumption in pregnancy and lactation. Nutrients 2018, 10, 1229.
- Hsu, C.N.; Chang-Chien, G.P.; Lin, S.; Hou, C.Y.; Tain, Y.L. Targeting on gut microbial metabolite trimethylamine-N-Oxide and short-chain fatty acid to prevent maternal high-fructose-diet-induced developmental programming of hypertension in adult male offspring. Mol. Nutr. Food Res. 2019, 63, e1900073.
- Tain, Y.L.; Lee, W.C.; Wu, K.L.H.; Leu, S.; Chan, J.Y.H. Resveratrol prevents the development of hypertension programmed by maternal plus post-weaning high-fructose consumption through modulation of oxidative stress, nutrient-sensing signals, and gut microbiota. Mol. Nutr. Food Res. 2018, 62, e1800066.
- Hsu, C.N.; Chan, J.Y.H.; Yu, H.R.; Lee, W.C.; Wu, K.L.H.; Chang-Chien, G.P.; Lin, S.; Hou, C.Y.; Tain, Y.L. Targeting on gut microbiota-derived metabolite trimethylamine to protect adult male rat offspring against hypertension programmed by combined maternal high-fructose intake and dioxin exposure. Int. J. Mol. Sci. 2020, 21, 5488.
- Hsu, C.N.; Hou, C.Y.; Chang-Chien, G.P.; Lin, S.; Yang, H.W.; Tain, Y.L. Perinatal resveratrol therapy prevents hypertension programmed by maternal chronic kidney disease in adult male offspring: Implications of the gut microbiome and their metabolites. Biomedicines 2020, 8, 567.
- Hsu, C.N.; Chan, J.Y.H.; Wu, K.L.H.; Yu, H.R.; Lee, W.C.; Hou, C.Y.; Tain, Y.L. Altered gut microbiota and its metabolites in hypertension of developmental origins: Exploring differences between fructose and antibiotics exposure. Int. J. Mol. Sci. 2021, 22, 2674.
- Hsu, C.N.; Hou, C.Y.; Chang-Chien, G.P.; Lin, S.; Chan, J.Y.H.; Lee, C.T.; Tain, Y.L. Maternal resveratrol therapy protected adult rat offspring against hypertension programmed by combined exposures to asymmetric dimethylarginine and trimethylamine-N-oxide. J. Nutr. Biochem. 2021, 93, 108630.
- Hsu, C.N.; Hou, C.Y.; Chang-Chien, G.P.; Lin, S.; Tain, Y.L. Maternal N-acetylcysteine therapy prevents hypertension in spontaneously hypertensive rat offspring: Implications of hydrogen sulfide-generating pathway and gut microbiota. Antioxidants 2020, 9, 856.
- Li, H.B.; Yang, T.; Richards, E.M.; Pepine, C.J.; Raizada, M.K. Maternal Treatment with captopril persistently alters gut-brain communication and attenuates hypertension of male offspring. Hypertension 2020, 75, 1315–1324.
- Wang, M.; Zhang, Y.; Miller, D.; Rehman, N.O.; Cheng, X.; Yeo, J.Y.; Joe, B.; Hill, J.W. Microbial reconstitution reverses early female puberty induced by maternal high-fat diet during lactation. Endocrinology 2020, 161, bqz041.
- Wankhade, U.D.; Zhong, Y.; Kang, P.; Alfaro, M.; Chintapalli, S.V.; Thakali, K.M.; Shankar, K. Enhanced offspring predisposition to steatohepatitis with maternal high-fat diet is associated with epigenetic and microbiome alterations. PLoS ONE 2017, 12, e0175675.
- Guimarães, K.S.L.; Braga, V.A.; Noronha, S.I.S.R.; Costa, W.K.A.D.; Makki, K.; Cruz, J.C.; Brandão, L.R.; Chianca Junior, D.A.; Meugnier, E.; Leulier, F.; et al. Lactiplantibacillus plantarum WJL administration during pregnancy and lactation improves lipid profile, insulin sensitivity and gut microbiota diversity in dyslipidemic dams and protects male offspring against cardiovascular dysfunction in later life. Food Funct. 2020, 11, 8939–8950.
- Hsu, C.N.; Hou, C.Y.; Lee, C.T.; Chan, J.Y.H.; Tain, Y.L. The Interplay between maternal and post-weaning high-fat diet and gut microbiota in the developmental programming of hypertension. Nutrients 2019, 11, 1982.
- Hsu, C.N.; Hou, C.; Chan, J.Y.H.; Lee, C.T.; Tain, Y.L. Hypertension programmed by perinatal high-fat diet: Effect of maternal gut microbiota-targeted therapy. Nutrients 2019, 11, 2908.
- Chen, H.E.; Lin, Y.J.; Lin, I.C.; Yu, H.R.; Sheen, J.M.; Tsai, C.C.; Huang, L.T.; Tain, Y.L. Resveratrol prevents combined prenatal NG-nitro-L-arginine-methyl ester (L-NAME) treatment plus postnatal high-fat diet induced programmed hypertension in adult rat offspring: Interplay between nutrient-sensing signals, oxidative stress and gut microbiota. J. Nutr. Biochem. 2019, 70, 28–37.
- Friedman, J.E.; Dobrinskikh, E.; Alfonso-Garcia, A.; Fast, A.; Janssen, R.C.; Soderborg, T.K.; Anderson, A.L.; Reisz, J.A.; D’Alessandro, A.; Frank, D.N.; et al. Pyrroloquinoline quinone prevents developmental programming of microbial dysbiosis and macrophage polarization to attenuate liver fibrosis in offspring of obese mice. Hepatol. Commun. 2018, 2, 313–328.
- de Oliveira, Y.; Cavalcante, R.G.S.; Cavalcanti Neto, M.P.; Magnani, M.; Braga, V.A.; de Souza, E.L.; de Brito Alves, J.L. Oral administration of Lactobacillus fermentum post-weaning improves the lipid profile and autonomic dysfunction in rat offspring exposed to maternal dyslipidemia. Food Funct. 2020, 11, 5581–5594.
- Sherman, S.B.; Sarsour, N.; Salehi, M.; Schroering, A.; Mell, B.; Joe, B.; Hill, J.W. Prenatal androgen exposure causes hypertension and gut microbiota dysbiosis. Gut Microbes 2018, 9, 400–421.
- Marzullo, P.; Di Renzo, L.; Pugliese, G.; De Siena, M.; Barrea, L.; Muscogiuri, G.; Colao, A.; Savastano, S. Obesity Programs of nutrition, Education, Research and Assessment (OPERA) Group. From obesity through gut microbiota to cardiovascular diseases: A dangerous journey. Int. J. Obes. Suppl. 2020, 10, 35–49.
- McMullen, S.; Mostyn, A. Animal models for the study of the developmental origins of health and disease. Proc. Nutr. Soc. 2009, 68, 306–320.
- Hsu, C.N.; Tain, Y.L. Animal models for DOHaD research: Focus on hypertension of developmental origins. Biomedicines 2021, 9, 623.
- Khodor, S.A.; Reichert, B.; Shatat, I.F. The microbiome and blood pressure: Can microbes regulate our blood pressure? Front. Pediatr. 2017, 5, 138.
- Yang, T.; Richards, E.M.; Pepine, C.J.; Raizada, M.K. The gut microbiota and the brain-gut-kidney axis in hypertension and chronic kidney disease. Nat. Rev. Nephrol. 2018, 14, 442–456.
- Pluznick, J.L. Microbial short-chain fatty acids and blood pressure regulation. Curr. Hypertens. Rep. 2017, 19, 25.
- Yang, T.; Santisteban, M.M.; Rodriguez, V.; Li, E.; Ahmari, N.; Carvajal, J.M.; Zadeh, M.; Gong, M.; Qi, Y.; Zubcevic, J.; et al. Gut dysbiosis is linked to hypertension. Hypertension 2015, 65, 1331–1340.
- Pluznick, J.L.; Protzko, R.J.; Gevorgyan, H.; Peterlin, Z.; Sipos, A.; Han, J.; Brunet, I.; Wan, L.X.; Rey, F.; Wang, T.; et al. Olfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proc. Natl. Acad. Sci. USA 2013, 110, 4410–4415.
- Ratajczak, W.; Rył, A.; Mizerski, A.; Walczakiewicz, K.; Sipak, O.; Laszczyńska, M. Immunomodulatory potential of gut microbiome-derived short-chain fatty acids (SCFAs). Acta Biochim. Pol. 2019, 66, 1–12.
- Hsu, C.N.; Hou, C.Y.; Chang-Chien, G.P.; Lin, S.; Tain, Y.L. Maternal garlic oil supplementation prevents high-fat diet-induced hypertension in adult rat offspring: Implications of H2S-generating pathway in the gut and kidneys. Mol. Nutr. Food Res. 2021, e2001116.
- Marques, F.Z.; Nelson, E.; Chu, P.-Y.; Horlock, D.; Fiedler, A.; Ziemann, M.; Tan, J.K.; Kuruppu, S.; Rajapakse, N.W.; El-Osta, A.; et al. High-fiber diet and acetate supplementation change the gut microbiota and prevent the development of hypertension and heart failure in hypertensive mice. Circulation 2017, 135, 964–977.
- Wang, Z.; Zhao, Y. Gut microbiota derived metabolites in cardiovascular health and disease. Protein Cell 2018, 9, 416–431.
- Seldin, M.M.; Meng, Y.; Qi, H.; Zhu, W.; Wang, Z.; Hazen, S.L.; Lusis, A.J.; Shih, D.M. Trimethylamine N-Oxide promotes vascular inflammation through signaling of mitogen-activated protein kinase and nuclear factor-κB. J. Am. Heart Assoc. 2016, 5, e002767.
- Leong, S.C.; Sirich, T.L. Indoxyl sulfate-review of toxicity and therapeutic strategies. Toxins 2016, 8, 358.
- Kamiński, T.W.; Pawlak, K.; Karbowska, M.; Myśliwiec, M.; Pawlak, D. Indoxyl sulfate-the uremic toxin linking hemostatic system disturbances with the prevalence of cardiovascular disease in patients with chronic kidney disease. BMC Nephrol. 2017, 18, 35.
- Hung, S.C.; Kuo, K.L.; Wu, C.C.; Tarng, D.C. Indoxyl sulfate: A novel cardiovascular risk factor in chronic kidney disease. J. Am. Heart Assoc. 2017, 6, e005022.
- Hubbard, T.D.; Murray, I.A.; Perdew, G.H. Indole and tryptophan metabolism: Endogenous and dietary routes to ah receptor activation. Drug Metab. Dispos. 2015, 43, 1522–1535.
- Zhang, N. The role of endogenous aryl hydrocarbon receptor signaling in cardiovascular physiology. J. Cardiovasc. Dis. Res. 2011, 2, 91–95.
- Wilck, N.; Matus, M.G.; Kearney, S.M.; Olesen, S.W.; Forslund, K.; Bartolomaeus, H.; Haase, S.; Mähler, A.; Balogh, A.; Markó, L.; et al. Salt-responsive gut commensal modulates TH17 axis and disease. Nature 2017, 551, 585–589.
- Brito, J.S.; Borges, N.A.; Esgalhado, M.; Magliano, D.C.; Soulage, C.O.; Mafra, D. Aryl hydrocarbon receptor activation in chronic kidney disease: Role of uremic toxins. Nephron 2017, 137, 1–7.
- Ren, J.; Crowley, S.D. Role of T-cell activation in salt-sensitive hypertension. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H1345–H1353.
- Hsu, C.N.; Lin, I.C.; Yu, H.R.; Huang, L.T.; Tiao, M.M.; Tain, Y.L. Maternal tryptophan supplementation protects adult rat offspring against hypertension programmed by maternal chronic kidney disease: Implication of tryptophan-metabolizing microbiome and aryl hydrocarbon receptor. Int. J. Mol. Sci. 2020, 21, 4552.
- Hsu, C.N.; Lin, Y.J.; Tain, Y.L. Maternal exposure to bisphenol a combined with high-fat diet-induced programmed hypertension in adult male rat offspring: Effects of resveratrol. Int. J. Mol. Sci. 2019, 20, 4382.
- Hsu, C.N.; Lin, Y.J.; Lu, P.C.; Tain, Y.L. Maternal resveratrol therapy protects male rat offspring against programmed hypertension induced by tcdd and dexamethasone exposures: Is it relevant to aryl hydrocarbon receptor? Int. J. Mol. Sci. 2018, 19, 2459.
- Kroll, M.H. The AHR: Adaptive evolution or one-off? Blood 2019, 134, 2337–2338.
- Karbowska, M.; Kaminski, T.W.; Znorko, B.; Domaniewski, T.; Misztal, T.; Rusak, T.; Pryczynicz, A.; Guzinska-Ustymowicz, K.; Pawlak, K.; Pawlak, D. Indoxyl sulfate promotes arterial thrombosis in rat model via increased levels of complex TF/VII, PAI-1, platelet activation as well as decreased contents of SIRT1 and SIRT3. Front. Physiol. 2018, 9, 1623.
- Nagareddy, P.; Smyth, S.S. Inflammation and thrombosis in cardiovascular disease. Curr. Opin. Hematol. 2013, 20, 457–463.
- Gryp, T.; Vanholder, R.; Vaneechoutte, M.; Glorieux, G. p-Cresyl Sulfate. Toxins 2017, 9, 52.
- Jing, Y.J.; Ni, J.W.; Ding, F.H.; Fang, Y.H.; Wang, X.Q.; Wang, H.B.; Chen, X.N.; Chen, N.; Zhan, W.W.; Lu, L.; et al. p-Cresyl sulfate is associated with carotid arteriosclerosis in hemodialysis patients and promotes atherogenesis in apoE-/- mice. Kidney Int. 2016, 89, 439–449.
- Hsu, C.-N.; Tain, Y.-L. Targeting the renin–angiotensin–aldosterone system to prevent hypertension and kidney disease of developmental origins. Int. J. Mol. Sci. 2021, 22, 2298.
- Paz Ocaranza, M.; Riquelme, J.A.; García, L.; Jalil, J.E.; Chiong, M.; Santos, R.A.S.; Lavandero, S. Counter-regulatory renin-angiotensin system in cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 116–129.
- Hsu, C.N.; Wu, K.L.; Lee, W.C.; Leu, S.; Chan, J.Y.; Tain, Y.L. Aliskiren administration during early postnatal life sex-specifically alleviates hypertension programmed by maternal high fructose consumption. Front. Physiol. 2016, 7, 299.
- Manning, J.; Vehaskari, V.M. Postnatal modulation of prenatally programmed hypertension by dietary Na and ACE inhibition. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R80–R84.
- Sherman, R.C.; Langley-Evans, S.C. Early administration of angiotensin-converting enzyme inhibitor captopril prevents the development of hypertension programmed by intrauterine exposure to a maternal low-protein diet in the rat. Clin. Sci. (Lond.) 1998, 94, 373–381.
- Oliveira Andrade, J.M.; de Farias Lelis, D.; Mafra, V.; Cota, J. The angiotensin converting enzyme 2 (ACE2), gut microbiota, and cardiovascular health. Protein Pept. Lett. 2017, 24, 827–832.
- Bessa, A.S.M.; Jesus, É.F.; Nunes, A.D.C.; Pontes, C.N.R.; Lacerda, I.S.; Costa, J.M.; Souza, E.J.; Lino-Júnior, R.S.; Biancardi, M.F.; Dos Santos, F.C.A.; et al. Stimulation of the ACE2/Ang-(1-7)/Mas axis in hypertensive pregnant rats attenuates cardiovascular dysfunction in adult male offspring. Hypertens. Res. 2019, 42, 1883–1893.
- Rubak, Y.T.; Nuraida, L.; Iswantini, D.; Prangdimurti, E. Angiotensin-I-converting enzyme inhibitory peptides in milk fermented by indigenous lactic acid bacteria. Vet. World 2020, 13, 345–353.
- Richards, E.M.; Pepine, C.J.; Raizada, M.K.; Kim, S. The gut, its microbiome, and hypertension. Curr. Hypertens. Rep. 2017, 19, 36.