Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Ava Boutilier and Version 2 by Rita Xu.
The M1/M2 macrophage paradigm plays a key role in tumor progression. M1 macrophages are historically regarded as anti-tumor, while M2-polarized macrophages, commonly deemed tumor-associated macrophages (TAMs), are contributors to many pro-tumorigenic outcomes in cancer through angiogenic and lymphangiogenic regulation, immune suppression, hypoxia induction, tumor cell proliferation, and metastasis.
- tumor-associated macrophage
- tumor microenvironment
- metastasis
- cytokine signaling
- polarization
- malignancy
1. Introduction
Macrophages are myeloid cells that are essential members of the innate immune response [1]. These heterogenous cells originate from monocyte precursors in the blood and differentiate in the presence of cytokines and growth factors in the tissues they infiltrate [2][3][2,3]. Macrophages are found in every human tissue in the body and exhibit anatomical and functional diversity [4]. These cells have three key functions: phagocytosis, exogenous antigen presentation, and immunomodulation through cytokine and growth factor secretion. As one of the earliest immune cells to encounter antigens at the site of infection or injury, their response is critical to the remainder of the immune response. Antigen-presentation, the ability to phagocytose, digest, and present antigen through the major histocompatibility complex class II (MHC II) system, is crucial for the activation of the adaptive immune system and highlights one of the key roles that macrophages play in the immune response [5]. T-cells (specifically T-helper cells) can recognize these MHC II complexes on classically activated macrophages through their T-cell receptors (TCRs), leading to further activation of the adaptive immune response [5]. Through phagocytosis, macrophages are also able to assist in the resolution of inflammation by effectively eliminating pathogen materials as well as assist in the clearance of apoptotic host cells such as neutrophils [6]. Without macrophages at the site of infection/tissue damage, an elevated apoptotic neutrophil population and elevated and prolonged inflammation has been demonstrated [7]. Macrophages are not only responsible for phagocytosis of foreign antigen; they also coordinate processes that initiate new tissue formation of the extracellular matrix and new blood vessel formation through angiogenesis under normal physiological conditions [4][8][4,8]. Macrophages, therefore, play a key role in tissue homeostasis under normal physiological conditions as well as after tissue damage. Macrophages also make key hematopoietic decisions by engulfing cells exuded from the bone marrow that do not express the CD47 ligand [9]. Macrophages engulf dead cells after infection or injury through recognition of the phosphatidylserine that is externalized on apoptotic cells. Phosphatidylserine is typically confined to the inner plasma membrane, but during apoptosis, it becomes externalized on the cell surface [10]. The engulfment of neutrophils and erythrocytes in the spleen and liver resolves the problems of neutropenia, splenomegaly, and reduced body weight [6].
Macrophages also regulate angiogenesis through different mechanisms. Macrophages can identify vascular endothelial cells and instruct them to undergo apoptosis if they do not receive countersignals from pericytes to survive. Macrophages have been shown to instruct functional angiogenesis in normal vessel maturation [11], wound healing [12], and development [13] and nonfunctional angiogenesis in many types of cancer [14] and chronic inflammatory conditions [15]. The protein, Wingless-Type MMTV Integration Site Family Member 7B (WNT7B), is secreted by macrophages and triggers apoptosis of vascular endothelial cells and, in the absence of WNT7B-secreting macrophages, there is significant vascular overgrowth. WNT7B secretion by macrophages is triggered by the presence of ANG2 secreted by mature blood vessels [13]. ANG2 secretion by blood vessels causes macrophages to upregulate WNT7B, which induces the proliferation of vascular endothelial cells and allows them to be targeted by ANG-2-induced apoptosis [13]. In response to Wnt7b, vascular endothelial cells enter the cell cycle and die in the G1 phase due to ANG2-mediated withdrawal of survival signals [16]. To induce angiogenesis, macrophages secrete WNT11 and WNT5A, which induce the expression of soluble vascular endothelial growth factor (VEGF) receptor 1 (VEGFR1) in an autocrine fashion. Soluble VEGFR1 regulates the levels of VEGF so vascular complexity is reduced, and the vasculature is more properly organized [17]. Wnt5a and Wnt11 are associated with non-canonical Wnt signaling. In a study of angiogenesis regulation by the non-canonical Wnt–Flt1 pathway, soluble and membrane-tethered VEGFR1 was measured by quantitative PCR in the RAW264.7 myeloid cell line after Wnt treatment, and both soluble and membrane-tethered VEGFR1 expression were significantly increased after RAW264.7 treatment with Wnt5a [17]. Macrophages are not limited to just blood vessel formation, but also play a key role in lymphangiogenesis during development and inflammation [18]. Macrophages can promote lymphangiogenesis by transdifferentiating into lymphatic endothelial cells and becoming incorporated into growing lymphatic vessels or by secreting growth factors and proteases that lead to lymphatic vessel formation [18]. Lymphangiogenesis can lead to tumor metastasis [19], making the role of macrophage regulation important to understand and further investigate.
2. Macrophage Markers
Regardless of origin, the major lineage regulator of almost all macrophages is the macrophage colony-stimulating factor 1 receptor in both humans (CSF1R) and mice (Csf1r). This is a class III, transmembrane tyrosine kinase receptor that is expressed on most mononuclear phagocytic cells [4]. While CSF1R/Csf1r is important in macrophage differentiation, Csf1r−/− mice have still exhibited some tissue macrophages, indicating the importance of other macrophage growth factors such as granulocyte–macrophage colony-stimulating factor (GM-CSF) and IL-3, which act as macrophage growth factors in tissue culture [4]. Macrophages also constitutively express the surface marker F4/80 in mice [4][13][4,13] and the equivalent constitutive markers CD14 and CD68 in humans [20][21][22][23][20,21,22,23].
2.1. Human Macrophage Markers
In humans, M1 macrophages are typically identified by the surface markers CD86 and CD64 [23][24][23,24]; the macrophage receptor with collagenous structure MARCO [23][25][26][23,25,26]; C-X-C chemokine ligand (CXCL) 9, 10, 11 (CXCL9, 10, 11) [23]; nitric oxide synthase 2 (NOS2), the suppressor of cytokine signaling 1 (SOCS1); and secretion of IL-6, IL-12, IL-1α [27], and TNF-α [24]. M2 macrophages typically express the surface markers CD206 [23][24][25][28][29][23,24,25,28,29] and CD163 [23][29][23,29] and express/secrete transforming growth factor-beta (TGF-β), peroxisome proliferator-activated receptor gamma (PPARγ), C-C motif chemokine ligands 14 and 22 (CCL14 and CCL22) [28], and arginase-1 (ARG-1) (Table 1) [18][22][18,22].
Table 1.
Macrophage polarization markers on M0, M1 and M2 mouse and human macrophages.
| Species | M0 | M1 | M2 |
|---|
| Mouse | Csf1r, F4/80, CD11b | Marco, Cxcl9, Cxcl10, Cxcl11, Nos2, Socs1 | Cd206, Tgm2, Fizz1, Chil3, Arg1, Ccl22, Cd163 |
| Human | CSF1R, CD14, CD68, CD11B | CD86, MARCO, CXCL9, CXCL10, CXCL11, NOS2, SOCS1, CD64 | TGM2, CD23, ARG1, CCL22, CD163, CD206 |
2.2. Mouse Macrophage Markers
While most macrophage markers are the same in both humans and mice, some exceptions exist. Murine M1 macrophages express macrophage receptors with collagenous structures (Marco) [23][25][26][23,25,26], Cxcl9, Cxcl10, Cxcl11 [23], Nos2, and Socs1, and secrete Il-6, Il-12, Il-1α [27], and Tnf-α [24], all comparable to their human counterparts. Murine M1 macrophages do not express surface markers CD86 or CD64. Murine M2 macrophages typically express the surface markers Cd206 [17][18][19][21][22][17,18,19,21,22] and Cd163 [23][29][23,29] and express/secrete Tgf-β, Pparγ, Ccl14 and Ccl22 [28], and Arg-1 [24][29][24,29], similar to human M2 macrophages; however, they also have Chitinase-3-like protein 3 (Chil3), unique from human M2 macrophages (Table 1) [29].
3. Macrophage Polarization
Many phenotypes of macrophages have been characterized based on their in vitro characteristics in cell culture experiments. Primarily, the classically activated M1 phenotype and the alternatively activated M2 phenotype are differentiated based on different surface receptor expression, secretory profiles, and functions [4][30][4,30]. Recent studies of gene expression of in vivo wound healing have shown that macrophages exhibit a pro-inflammatory M1 secretory profile during the early stages and then transition to an anti-inflammatory M2 gene expression profile during the later healing stages [31]. Macrophage polarization refers to the activation state of a macrophage at a singular point in time, but due to the plasticity of macrophages, their polarization state is not fixed and can be altered based on the integration of multiple signals from other cells, tissues, and pathogens [32]. While macrophage polarization is typically discussed as a singular point in time, it is important to understand that M2-type macrophages can switch to an M1 phenotype, or vice versa, based on environmental changes such as cytokine and growth factor secretion, inflammation, infection, injury, hypoxia, and other conditions. Macrophage polarization is more complex than the M1 and M2 binary classification, with those subtypes representing the extremes on the spectrum of macrophage polarization (Figure 1). Many of these subsets express combinations of M1 and M2 cell markers and have yet to be formally defined.
Figure 1. Macrophage polarization gradient. This figure illustrates the heterogeneity of macrophage polarization in place of binary M1/M2 classifications. The pro-inflammatory M1 and anti-inflammatory M2 cells lie on opposite ends of the polarization axis, but many macrophages with mixed pro- and anti-inflammatory characteristics exist in between. Environmental changes may cause macrophages to shift from M1 to M2, vice versa, or to a hybrid of both cells. This highlights the plasticity of macrophages and interdependence on the surrounding environment. This figure was created with Biorender.com (accessed on 1 May 2021).
3.1. M1 and M2 Macrophages
M1 macrophages are pro-inflammatory in nature and are characterized by their high capacity to present antigens, produce interleukin 12 and 23 (IL-12 and IL-23) [33], and activate type-I T-cell responses [5]. They inhibit cell proliferation and cause tissue damage through the secretion of pro-inflammatory cytokines and nitric oxide (NO) and are induced by T-helper type-1 cytokines including interferon-γ (IFN-γ), interleukin-1β (IL-1β), and lipopolysaccharide (LPS) [1][3][4][34][1,3,4,34]. M2 macrophages are typically anti-inflammatory in nature and are characterized by their poor ability to present antigen; having low IL-12 and high IL-10, IL-4, and IL-13 secretory profiles; and immunosuppressive effects [5]. These cells promote cell proliferation, tissue repair, angiogenesis, and phagocytosis to downregulate inflammation and “clean up” after inflammatory events and are T-helper type-2 activators and TH1 inhibitors [1][3][4][5][34][1,3,4,5,34]. While macrophage polarization is often defined as a specific moment in time, it is important to note that these markers are often present on many subtypes of macrophages in varying expression levels. For example, M2 macrophages can still express M1 markers but with lower levels than M1 macrophages, and vice versa [35].
3.2. Extrinsic Polarization
Extrinsic polarization is a primary method of macrophage polarization and is mediated by cytokine secretion by other cells such as CD4+ TH1 or TH2 cells (Table 2). Some non-cytokine, extrinsic pathways of macrophage polarization do exist, however, including hypoxia as well as the production of lactate within tumors, which drive M2 polarization [36].
Table 2. Overview of extrinsic mechanisms of macrophage polarization.
| Protein/Gene | Normal Function | Effect on Polarization |
|---|---|---|
| Interleukin-4 and Interleukin-13 | Cytokines | M2-favored |
| Interleukin-4 receptor alpha | IL-4 and IL-13 signaling | M2-favored |
| Signal transducer and activator of transcription 6 | Transcription factor | M2-favored |
| Peroxisome proliferator activated receptor gamma | Transcription factor | M2-favored |
| Tubular sclerosis 1 | Inhibitor of mTOR | M2-favored |
| AKT Serine/Threonine Kinase 1 | Signaling | M2-favored |
| AKT Serine/Threonine Kinase 2 | Signaling | M1-favored |
| Src homology region 2 domain-containing phosphatase-1/2 | Phosphatases | M1-favored |
| SH2-containing Inositol 5′-Phosphatase | Phosphatase | M1-favored |
| Phosphatase and tensin homolog | Lipid phosphatase | M1-favored |
| Myeloid differentiation primary response 88 | Signaling adapter | M1-favored |
| Tumor necrosis factor | Cytokine | M1-favored |
| Tumor necrosis factor receptor 1 | Cytokine receptor | M1-favored |
| Interferon-gamma, Interleukin-12 | Cytokines | M1-favored |
To detail Table 2 and Figure 2, TH1 cells secrete IFN-γ, which drives polarization of macrophages towards an M1 phenotype, while TH2 cells secrete IL-4 and IL-13, which drive M2-phenotype polarization [34][37][34,37]. IL-4 and IL-13 inhibit the production of nitric oxide, an inflammatory mediator, through the depletion of arginine, which serves as the substrate for iNOS/Nos2. This inhibition of NO production in macrophages was found to be dependent on IL-4 or IL-13 through the depletion of Arg-1 through a Stat6-dependent pathway [38]. This inhibition of NO production leads to a loss of the M1 phenotype and polarization toward the M2 phenotype through cytokines IL-4 and IL-13. IL-4 and LPS signaling can also target the mechanistic target of rapamycin (mTOR) and Akt to trigger polarization. In LPS-mediated M1 polarization, Toll-like receptor 4 (TLR4) activates phosphoinositide 3-kinase (PI3K) followed by Akt and mammalian target of rapamycin complex 1 (mTORC1) activation, leading to M1 polarization [39][40][39,40]. Additionally, pharmacological and genetic inhibition of Akt1/2 has shown that Akt1 inhibits M1 activation, and Akt2 leads to the activation of M1 genes, favoring polarization to the M1 phenotype [39][40][39,40]. Akt signaling is likely to control macrophage polarization through downstream effectors; for example, Akt signaling inhibits transcription factor Foxo1, which is a key gene in M1 macrophages. Additionally, Akt1 has been implicated as a negative regulator of the nuclear factor, kappa-light-chain-enhanced activity of activated B cells (NF-κB), while Akt2 is a positive regulator. NF-κB is a master regulator of M1 activation [41]. Src homology region 2 domain-containing phosphatase 1/2 (SHP-1/2) inhibits CD11b activity, therefore inhibiting M2 polarization and leading to an increase in M1-type macrophages [42]. Src homology 2 (SH2) domain-containing inositol polyphosphate 5-phosphatase (SHIP) is another phosphatase that inhibits the activation of M2-like macrophages. SHIP−/− peritoneal and alveolar macrophages have been found to be profoundly M2-skewed, with high arginase I levels and impaired LPS-stimulated NO production [43]. Phosphatase and tensin homolog (PTEN) plays a key role in regulating the inflammatory response through M1-polarization. Mice with a myeloid-specific PTEN knockout have been shown to have levels of M2 macrophages and produce lower TNF-α and higher IL-10 in response to TLR ligands, indicating that PTEN plays a key role in M1 macrophage differentiation [44]. Tumor necrosis factor (TNF) has been found to be a positive regulator of M1 polarization through its activation of the NF-κB pathway. Tumor necrosis factor receptor (TNFR) signaling was found to be a negative regulator of M2 polarization in tumor-associated macrophages (TAMs), and myeloid differentiation primary response 88 (MyD88) was shown to suppress M2 gene expression in TAMs, leading to an M1 phenotype [45].
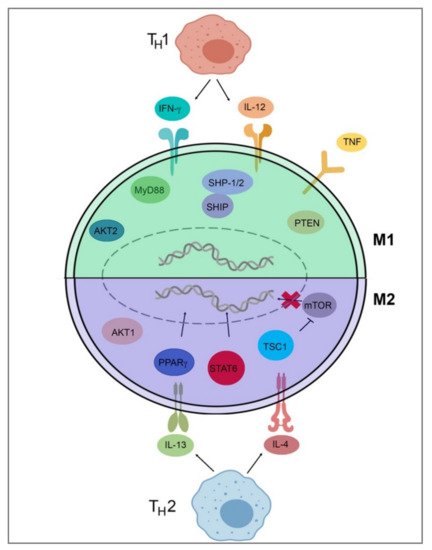
Figure 2. Signal pathways of macrophage polarization. This figure illustrates several of the various mechanisms that drive extrinsic macrophage polarization. Those pathways include IFN-y and IL-12 secretion by TH1 T-cells, LPS signaling through mTOR/Akt or TLR4, Akt2/NF-κB activation, SHP-1/2 inhibition of Cd11b, SHIP and MyD88 inhibition of M2 genes, PTEN activation, and TNF/TNFR/NF-κB activation to induce M1 gene expression. Induction of M2 genes is directed by the secretion of IL-4 and IL-13 from TH2 T-cells and IL-4Rα receptor activation as well as downstream Stat6-dependent arginase-1 inhibition, PPARγ activation, and TSC1 inhibition of mTOR. This figure was created with Biorender.com (accessed on 1 May 2021).