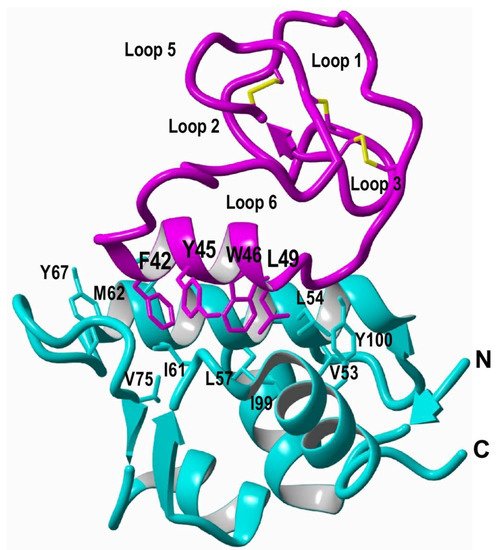
Cyclotides are a novel class of micro-proteins (≈30–40 residues long) with a unique topology containing a head-to-tail cyclized backbone structure further stabilized by three disulfide bonds that form a cystine knot. This unique molecular framework makes them exceptionally stable to physical, chemical, and biological degradation compared to linear peptides of similar size. The cyclotides are also highly tolerant to sequence variability, aside from the conserved residues forming the cystine knot, and are orally bioavailable and able to cross cellular membranes to modulate intracellular protein–protein interactions (PPIs), both in vitro and in vivo. These unique properties make them ideal scaffolds for many biotechnological applications, including drug discovery.
- cyclotides
- CCK
- cystine-knot
- drug design
- backbone cyclized polypeptides
- protein-protein interactions
- cyclic peptides
1. Introduction
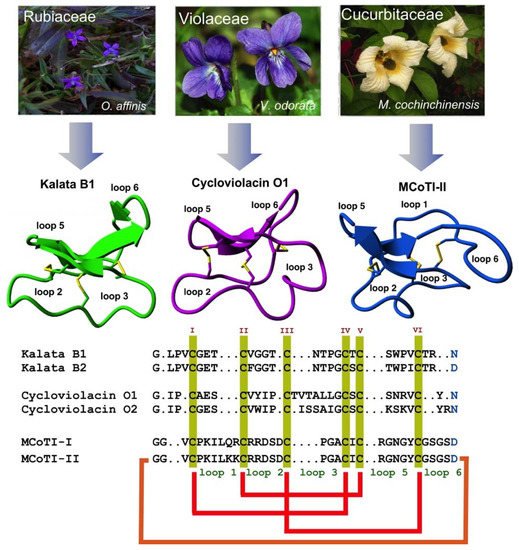
2. Structure
3. Biosynthesis
4. Chemical Synthesis
5. Recombinant Expression
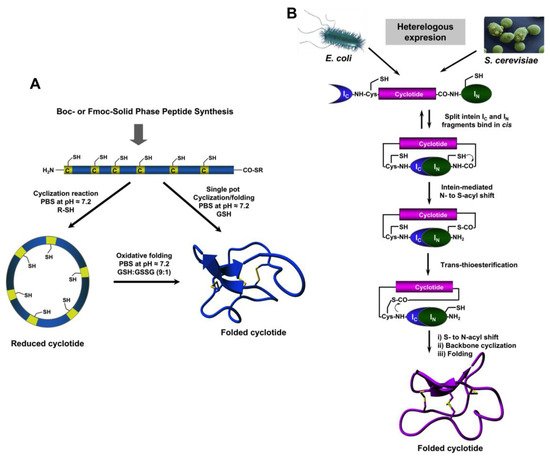
The use of protein splicing units, also called inteins, in either cis or trans allows the recombinant production of backbone cyclized polypeptides (for more detailed reviews in this topic see [27][73][27,70]). Initial attempts for production of cyclotides using heterologous expression systems involved the use of modified inteins for generating α-thioester polypeptides that were then backbone-cyclized using an intramolecular version of native chemical ligation [74][75][71,72]. The use of intein-mediated protein trans-splicing (PTS) has been shown to be more effective for the production of naturally-occurring and engineered cyclotides in prokaryotic and eukaryotic expression systems (Figure 2B) [67][68][69][73,74,75]. In-cell production of folded cyclotides by PTS can reach intracellular concentrations in the range of 2040– μM. This corresponds to ≈ 10 mg of folded cyclotide per 100 g of wet cells in Escherichia coli expression systems producing cyclotide MCoTI-I [69][75]. These values are quite comparable to those obtained when using the cyclotide-producing plant O. affinis, which produces ≈ 15 mg of cyclotide kalata B1 per 100 g of wet weight when grown in vitro [76]. Given the fastest growth rate and the simplicity of working with microorganisms such as E. coli, PTS provides a very attractive alternative for a cost-effective route to produce bioactive cyclotides with therapeutic potential. In-cell production of cyclotides also opens the exciting possibility for the generation of large genetically-encoded libraries of cyclotides, which can be rapidly screened for the selection of novel sequences able to modulate specific molecular targets [68][74]. In addition, having easy access to cyclotides using standard heterologous expression systems facilitates the production of cyclotides labeled with NMR active isotopes, such as 15N and 13C, in a relatively inexpensive fashion [5]. This approach was used to carry out structural studies using heteronuclear NMR on a cyclotide engineered to bind the p53 binding domain of the E3-ligases Hdm2 and HdmX, allowing elucidation of the structure of the bioactive cyclotide bound to its target (Figure 3) [5].