Neuromuscular diseases (NMDs) are a heterogeneous group of acquired or inherited rare disorders caused by injury or dysfunction of the anterior horn cells of the spinal cord (lower motor neurons), peripheral nerves, neuromuscular junctions, or skeletal muscles leading to muscle weakness and waste. Unfortunately, most of them entail serious or even fatal consequences. The prevalence rates among NMDs range between 1 and 10 per 100,000 population, but their rarity and diversity pose difficulties for healthcare and research. Some molecular hallmarks are being explored to elucidate the mechanisms triggering disease, to set the path for further advances. In fact, in the present review we outline the metabolic alterations of NMDs, mainly focusing on the role of mitochondria. The aim of the review is to discuss the mechanisms underlying energy production, oxidative stress generation, cell signaling, autophagy, and inflammation triggered or conditioned by the mitochondria. Briefly, increased levels of inflammation have been linked to reactive oxygen species (ROS) accumulation, which is key in mitochondrial genomic instability and mitochondrial respiratory chain (MRC) dysfunction. ROS burst, impaired autophagy, and increased inflammation are observed in many NMDs. Increasing knowledge of the etiology of NMDs will help to develop better diagnosis and treatments, eventually reducing the health and economic burden of NMDs for patients and healthcare systems.
- neuromuscular diseases (NMDs)
- mitophagy
- reactive oxygen species (ROS)
- damage-associated molecular patterns (DAMPs)
- oxidative stress
- mitochondrial respiratory chain (MRC)
1. Mitochondrial Genome and mtDNA Mutations
2. Mitochondrial Respiratory Chain (MRC)
3. Mitochondrial Quality Control
3.1. Mitochondrial Biogenesis
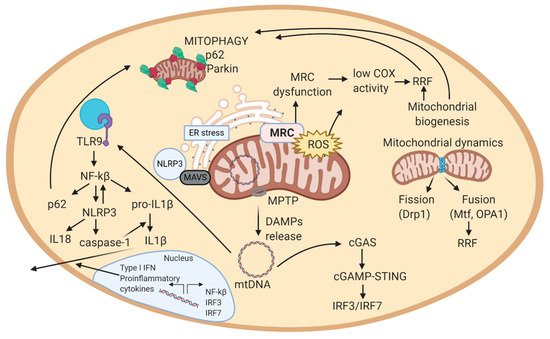
3.2. Mitochondrial Dynamics
3.3. Autophagy and Mitophagy
4. Mitochondrial ROS and ER Stress
5. Mitochondrially Induced Inflammatory Response
5.1. Implication of mtDNA in Inflammation
well. Mitochondria are suggested to regulate the activity of the NLRP3 inflammasome complex, by: activating mitophagy (reduces inflammation by clearing mitochondrial-bound NLRP3 complexes) and releasing mitochondrial ROS (amplifies inflammasome immunogenic signal) [140].
dominant optic atrophy [101]. This pathologic feature has been described in sIBM and in LMNA-related NMDs (CMD, Emery-Dreifuss MD, LGMD, CMT, etc.)) [144]. Further, cytosolic mtDNA can activate cGAS-STING to induce IFN-1 or the NLRP3 inflammasome which can induce the maturation/secretion of pro-inflammatory cytokines [101]. Thus, mtDNA can activate major innate immune responses by acting as a DAMP from the mitochondria. Overall, mtDNA is released from mitochondria to the cytosol, where it is recognized by immune receptors, which trigger a signaling cascade that leads to the production of cytokines or transcription of inflammatory genes in the nucleus (Figure 2) [143]. This close association between mitochondria and inflammation triggered from affected cells, explains why NMDs are usually associated with inflammatory effects.
5.2. Implication of Mitochondrial ROS in Inflammation
5.3. Feedback Regulation of the Mitochondrial Inflammatory Response
attenuates PAMP and DAMP availability [100].
critical for preventing disease development.
References
- Kauppila, T.E.S.; Kauppila, J.H.K.; Ran Larsson, N.-G. Cell Metabolism Review Mammalian Mitochondria and Aging: An Update. Cell Metab. 2017, 25, 57–71.
- Piantadosi, C.A.; Suliman, H.B. Transcriptional control of mitochondrial biogenesis and its interface with inflammatory processes. Biochim. Biophys. Acta 2012, 1820, 532–541.
- Couvillion, M.T.; Soto, I.C.; Shipkovenska, G.; Stirling, L. Synchronized mitochondrial and cytosolic translation programs. Nature 2016, 533, 499–503.
- Tokuyama, T.; Hirai, A.; Shiiba, I.; Ito, N.; Matsuno, K.; Takeda, K.; Saito, K.; Mii, K.; Matsushita, N.; Fukuda, T.; et al. Mitochondrial Dynamics Regulation in Skin Fibroblasts from Mitochondrial Disease Patients. Biomolecules 2020, 10, 450.
- Nardin, R.A.; Johns, D.R. Mitochondrial dysfunction and neuromuscular disease. Muscle Nerve 2001, 24, 170–191.
- Stewart, J.B.; Chinnery, P.F. The dynamics of mitochondrial DNA heteroplasmy: Implications for human health and disease. Nat. Rev. Genet. 2015, 16, 530–542.
- Picca, A.; Lezza, A.M.S.; Leeuwenburgh, C.; Pesce, V.; Calvani, R.; Landi, F.; Bernabei, R.; Marzetti, E. Fueling inflamm-aging through mitochondrial dysfunction: Mechanisms and molecular targets. Int. J. Mol. Sci. 2017, 18.
- Frank, M.; Duvezin-Caubet, S.; Koob, S.; Occhipinti, A.; Jagasia, R.; Petcherski, A.; Ruonala, M.O.; Priault, M.; Salin, B.; Reichert, A.S. Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 2297–2310.
- Babbar, M.; Basu, S.; Yang, B.; Croteau, D.L.; Bohr, V.A. Mitophagy and DNA damage signaling in human aging. Mech. Ageing Dev. 2020, 186.
- Siciliano, G.; Monzani, F.; Manca, M.L.; Tessa, A.; Caraccio, N.; Tozzi, G.; Piemonte, F.; Mancuso, M.; Santorelli, F.M.; Ferrannini, E.; et al. Human Mitochondrial Transcription Factor A Reduction and Mitochondrial Dysfunction in Hashimoto’s Hypothyroid Myopathy. Mol. Med. 2002, 8, 326–333.
- Bomont, P.; Cavalier, L.; Blondeau, F.; Hamida, C.B.; Belal, S.; Tazir, M.; Demir, E.; Topaloglu, H.; Korinthenberg, R.; Tüysüz, B.; et al. The gene encoding gigaxonin, a new member of the cytoskeletal BTB/kelch repeat family, is mutated in giant axonal neuropathy. Nat. Lett. 2000, 26, 370–374.
- Vincent, A.E.; Ng, Y.S.; White, K.; Davey, T.; Mannella, C.; Falkous, G.; Feeney, C.; Schaefer, A.M.; Mcfarland, R.; Gorman, G.S.; et al. The Spectrum of Mitochondrial Ultrastructural Defects in Mitochondrial Myopathy. Sci. Rep. 2016, 6, 30610.
- Bhatt, P.S.; Tzoulis, C.; Balafkan, N.; Miletic, H.; Tran, G.T.T.; Sanaker, P.S.; Bindoff, L.A. Mitochondrial DNA depletion in sporadic inclusion body myositis. Neuromuscul. Disord. 2019, 29, 242–246.
- De Duve, C.; Pressman, B.C.; Gianetto, R.; Wattiaux, R.; Appelmans, F. Tissue fractionation studies. Biochem. J. 1955, 60, 604–617.
- Kepp, O.; Galluzzi, L.; Kroemer, G. Mitochondrial control of the NLRP3 inflammasome. Nat. Immunol. 2011, 12.
- Katsetos, C.D.; Koutzaki, S.; Melvin, J.J. Mitochondrial dysfunction in neuromuscular disorders. Semin. Pediatric Neurol. 2013, 20, 202–215.
- Koopman, W.J.H.; Willems, P.H.G.M.; Smeitink, J.A.M. Monogenic Mitochondrial Diseases. N. Engl. J. Med. 2012, 366, 1132–1141.
- Keogh, M.J.; Chinnery, P.F. Mitochondrial DNA mutations in neurodegeneration. Biochim. Biophys. Acta Bioenerg. 2015, 1847, 1401–1411.
- Lightowlers, R.N.; Taylor, R.W.; Turnbull, D.M. Mutations causing mitochondrial disease: What is new and what challenges remain? Science 2015, 349, 1494–1499.
- Argov, Z.; Renshaw, P.F.; Boden, B.; Winokur, A.; Bank, W.J. Effects of thyroid hormones on skeletal muscle bioenergetics. In vivo phosphorus-31 magnetic resonance spectroscopy study of humans and rats. J. Clin. Investig. 1988, 81, 1695–1701.
- Monzani, F.; Caraccio, N.; Siciliano, G.; Manca, L.; Murri, L.; Ferrannini, E. Clinical and Biochemical Features of Muscle Dysfunction in Subclinical Hypothyroidism. J. Clin. Endocrinol. Metab. 1997, 82, 3315–3318.
- Deng, J.; Wang, P.; Chen, X.; Cheng, H.; Liu, J.; Fushimi, K.; Zhu, L.; Wu, J.Y. FUS interacts with ATP synthase beta subunit and induces mitochondrial unfolded protein response in cellular and animal models. Proc. Natl. Acad. Sci. USA 2018, 115, E9678–E9686.
- Spiegel, R.; Saada, A.; Flannery, P.J.; Burté, F.; Soiferman, D.; Khayat, M.; Eisner, V.; Vladovski, E.; Taylor, R.W.; Bindoff, L.A.; et al. Fatal infantile mitochondrial encephalomyopathy, hypertrophic cardiomyopathy and optic atrophy associated with a homozygous OPA1 mutation. J. Med. Genet. 2016, 53, 127–131.
- Rodríguez-Nuevo, A.; Díaz-Ramos, A.; Noguera, E.; Díaz-Sáez, F.; Duran, X.; Muñoz, J.P.; Romero, M.; Plana, N.; Sebastián, D.; Tezze, C.; et al. Mitochondrial DNA and TLR9 drive muscle inflammation upon Opa1 deficiency. EMBO J. 2018, 37.
- Züchner, S.; Mersiyanova, I.V.; Muglia, M.; Bissar-Tadmouri, N.; Rochelle, J.; Dadali, E.L.; Zappia, M.; Nelis, E.; Patitucci, A.; Senderek, J.; et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004, 36, 449–451.
- Mishra, P.; Carelli, V.; Manfredi, G.; Chan, D.C. Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. 2014, 19, 630–641.
- Waterham, H.R.; Koster, J.; van Roermund, C.W.T.; Mooyer, P.A.W.; Wanders, R.J.A.; Leonard, J.V. A Lethal Defect of Mitochondrial and Peroxisomal Fission. N. Engl. J. Med. 2007, 356, 1736–1741.
- Kodavati, M.; Wang, H.; Hegde, M.L. Altered Mitochondrial Dynamics in Motor Neuron Disease: An Emerging Perspective. Cells 2020, 9, 1065.
- Clark, S. Newborn mice studied with the electron mircroscope. Biophys. Biochem. Cytol. 1957, 3, 349–362.
- Ohsumi, Y. Historical landmarks of autophagy research. Cell Res. 2014, 24, 9–23.
- Eiyama, A.; Okamoto, K. PINK1/Parkin-mediated mitophagy in mammalian cells. Curr. Opin. Cell Biol. 2015, 33, 95–101.
- Seabright, A.P.; Fine, N.H.F.; Barlow, J.P.; Lord, S.O.; Musa, I.; Gray, A.; Bryant, J.A.; Banzhaf, M.; Lavery, G.G.; Hardie, D.G.; et al. AMPK activation induces mitophagy and promotes mitochondrial fission while activating TBK1 in a PINK1-Parkin independent manner. FASEB J. 2020, 34, 6284–6301.
- García-Prat, L.; Martínez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodríguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42.
- Lemasters, J.J. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005, 8, 3–5.
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14.
- Sun, N.; Yun, J.; Liu, J.; Malide, D.; Liu, C.; Rovira, I.I.; Holmström, K.M.; Fergusson, M.M.; Yoo, Y.H.; Combs, C.A.; et al. Measuring In Vivo Mitophagy. Mol. Cell 2015, 60, 685–696.
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nat. Lett. 2010, 464, 104–107.
- Grumati, P.; Grumati, P.; Coletto, L.; Sabatelli, P.; Cescon, M.; Angelin, A.; Bertaggia, E.; Blaauw, B.; Urciuolo, A.; Tiepolo, T.; et al. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat. Med. 2010, 16, 1313–1320.
- Zhang, W.; Hu, X.; Shen, Q.; Xing, D. Mitochondria-specific drug release and reactive oxygen species burst induced by polyprodrug nanoreactors can enhance chemotherapy. Nat. Commun. 2019, 10, 1–14.
- Lerner, C.A.; Sundar, I.K.; Rahman, I. Mitochondrial redox system, dynamics, and dysfunction in lung inflammaging and COPD. Int. J. Biochem. Cell Biol. 2016, 81, 294–306.
- Domènech, B.E.; Marfany, G. The Relevance of Oxidative Stress in the Pathogenesis and Therapy of Retinal Dystrophies. Antioxidants 2020, 9, 347.
- Peterman, E.M.; Sullivan, C.; Goody, M.F.; Rodriguez-Nunez, I.; Yoder, J.A.; Kim, C.H. Neutralization of mitochondrial superoxide by superoxide dismutase 2 promotes bacterial clearance and regulates phagocyte numbers in zebrafish. Infect. Immun. 2015, 83, 430–440.
- Andersen, P.M.; Nilsson, P.; Forsgren, L.; Marklund, S.L. CuZn-Superoxide Dismutase, Extracellular Superoxide Dismutase, and Glutathione Peroxidase in Blood from Individuals Homozygous for Asp90Ala CuZn-Superoxide Dismutase Mutation. J. Neurochem. 2002, 70, 715–720.
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 3460–3470.
- Manevski, M.; Muthumalage, T.; Devadoss, D.; Sundar, I.K.; Wang, Q.; Singh, K.P.; Unwalla, H.J.; Chand, H.S.; Rahman, I. Cellular stress responses and dysfunctional Mitochondrial–cellular senescence, and therapeutics in chronic respiratory diseases. Redox Biol. 2020, 33, 101443.
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933.
- Naon, D.; Scorrano, L. At the right distance: ER-mitochondria juxtaposition in cell life and death. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 2184–2194.
- Nadalutti, C.A.; Stefanick, D.F.; Zhao, M.-L.; Horton, J.K.; Prasad, R.; Brooks, A.M.; Griffith, J.D.; Wilson, S.H. Mitochondrial dysfunction and DNA damage accompany enhanced levels of formaldehyde in cultured primary human fibroblasts. Sci. Rep. 2020, 10, 5575.
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.K.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Pyo Kim, H.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 8, 222–230.
- Pérez-Treviño, P.; Velásquez, M.; García, N. Mechanisms of mitochondrial DNA escape and its relationship with different metabolic diseases. BBA Mol. Basis Dis. 2020, 1866, 165761.
- Rodríguez-Nuevo, A.; Zorzano, A. The sensing of mitochondrial DAMPs by non-immune cells. Cell Stress 2019, 3, 195–207.