Cardiovascular disease is the leading cause of morbidity and mortality in the western and developing world, and the incidence of cardiovascular disease is increasing with the longer lifespan afforded by our modern lifestyle. Vascular diseases including coronary heart disease, high blood pressure, and stroke comprise the majority of cardiovascular disease and therefore represent a significant medical and socioeconomic burden on our society. It is not be surprising that these conditions overlap and potentiate each other when we consider the many cellular and molecular similarities between them. At the molecular level, the vascular smooth muscle cell (VSMC) is the target, integrator, and effector cell of both atherogenic and the major effector protein of the hypertensive signal, Angiotensin II (Ang II). Together, these signals can potentiate each other and prime the artery and exacerbate hypertension and atherosclerosis. Therefore, VSMC are the fulcrum in progression of these diseases and therefore, understanding the effects of atherogenic stimuli and Ang II on VSMC is key to understanding and treating atherosclerosis and hypertension. In this review, we will examine studies in which hypertension and atherosclerosis intersect on the VSMC, and illustrate common pathways between these two diseases and vascular aging.
Cardiovascular disease is the leading cause of morbidity and mortality in the western and developing world, and the incidence of cardiovascular disease is increasing with the longer lifespan afforded by our modern lifestyle. Vascular diseases including coronary heart disease, high blood pressure, and stroke comprise the majority of cardiovascular disease and therefore represent a significant medical and socioeconomic burden on our society. It is not be surprising that these conditions overlap and potentiate each other when we consider the many cellular and molecular similarities between them. At the molecular level, the vascular smooth muscle cell (VSMC) is the target, integrator, and effector cell of both atherogenic and the major effector protein of the hypertensive signal, Angiotensin II (Ang II). Together, these signals can potentiate each other and prime the artery and exacerbate hypertension and atherosclerosis. Therefore, VSMC are the fulcrum in progression of these diseases and therefore, understanding the effects of atherogenic stimuli and Ang II on VSMC is key to understanding and treating atherosclerosis and hypertension. In this review, we will examine studies in which hypertension and atherosclerosis intersect on the VSMC, and illustrate common pathways between these two diseases and vascular aging.
- vascular smooth muscle cell
- Angiotensin II
- hypercholesterolemia
- atherosclerosis
- hypertension
- vascular disease
1
1. Definition
Vascular diseases including coronary heart disease, high blood pressure, and stroke comprise the majority of cardiovascular diseases, and therefore represent a significant medical and socioeconomic burden on our society. It may not be surprising that these conditions overlap and potentiate each other when we consider the many cellular and molecular similarities between them. These intersecting points are manifested in clinical studies in which lipid lowering therapies reduce blood pressure, and anti-hypertensive medications reduce atherosclerotic plaque. At the molecular level, the vascular smooth muscle cell (VSMC) is the target, integrator, and effector cell of both atherogenic and the major effector protein of the hypertensive signal Angiotensin II (Ang II). Together, these signals can potentiate each other and prime the artery and exacerbate hypertension and atherosclerosis. Therefore, VSMCs are the fulcrum in progression of these diseases and, therefore, understanding the effects of atherogenic stimuli and Ang II on the VSMC is key to understanding and treating atherosclerosis and hypertension.
2. Introduction
2.1. Hypertension and Angiotensin II
The renin-angiotensin system (RAS) functions in an endocrine fashion to influence numerous organs throughout the body. The DefinitRAS plays an essential role in the regulation of blood pressure through the regulation
V of peripheral vascular resistance andiseases including coronary heart disease, high blood pressure, and stro by influencing the electrolyte and fluid balance in the organism. The major effector peptide of this system, angiotensin II (Ang II), is produced from the conversion of angiotensinogen to angiotensin I by renin, which is then cleaved by angiotensin-converting enzyme (ACE). Ang II is recognized by two different G-protein-coupled receptors, Angiotensin type 1 (AT1R) and Angiotensin type 2 (AT2R). Angiotensin type 1 receptors are expressed in most tissue and mediate most of Ang II’s effects, particularly those that are vasoactive. Vascular type-1 and type-2 receptors have opposing effects. While AT1R mediates hypertensive effects, AT2R activation produces hypotension, and has anti-hypertensive effects. AT1R signaling and mediated gene expression will be the major focus of this review. While AT1R is widely expressed in many tissues, we will limit our discussion to that which is reported in VSMC. AT1R is a G-protein coupled receptor (GPCR), and upon ligand binding activates the classical G-protein related pathway. Canonical GPCR signaling regulates VSMC contractility in a calcium/calmodulin-mediated pathway, which activates MLCK activation and actin/myosin interactions to mechanize cellular contraction [1]. This receptor-mediated pathway also activates receptor tyrosine kinase comprise the majoris such as epidermal growth factor receptor (EGFR) and platelet derived growth factor receptor (PDGF), among others. Importantly for this review, AT1R activates p38, a MAPK and central integrator of inflammatory signaling [2]. Ang II stimulated MAPK activation is significantly of cardio higher in VSMC isolated and cultured from spontaneously hypertensive rats (SHR) compared to normal VSMC, suggesting an intimate relationship between the AT1R and MAPK pathways [3]. One distal event of p38 ascular diseasnd receptor tyrosine kinase activation is the activation of transcription factors, most-notably NF-κB, a master regulator of pro-inflammatory gene expression. In cultured VSMC and aorta of animals, Ang II directly activates NF-κB through AT1R [4]. It has long been recognized that in VSMC, Ang II is a powerful activator of oxidant stress, and therefore repa second major pathway through which Ang II activates NF-κB is through oxidative stress. Ang II generation of reactant oxygen species (ROS) in a NAD(P)H-dependent mechanism leads to a number of maladaptive cellular responses [5][6]. Although vascular NAD(P)H are esent a signsential in the normal physiology of VSMC, they also catalyze excessive ROS (for excellent review, see [7]). ROS in turn, by virtue oficant medical and socioecon being a potent second messenger, activates a number of transcription factors in VSMC, again most notably NF-κB. Pertinent in any discussion of atherosclerosis, this results in maladaptive expression of inflammatory cytokines such as TNFα, chemokines like MCP-1, adhesion molecules such as ICAM1, and matrix and matrix modifying proteinases [8][9][10]. Together, these mic burden on our society. It may not be surprising that these conolecular events act in concert and result in cellular responses such as VSMC hypertrophy, proliferation, oxidation, and altered matrix metabolism, all which lead to classic pathophysiological indices of vascular stiffness, loss of compliance, and remodeling indicative of hypertension. These molecular events are summarized in Figure 1. Importantly, this also leaves the vessel wall primed for inflammatory insult by hypercholesterolemia. Clinically, atherosclerosis and hypertension are considered to be separate diseases with different diagnoses and treatments. This being said, from a molecular, cellular and vascular perspective, there are many similarities, and perhaps too many opportunities for cross-talk between hypertensive and atherogenic stimuli and outcomes. These will be discussed in the next sections.
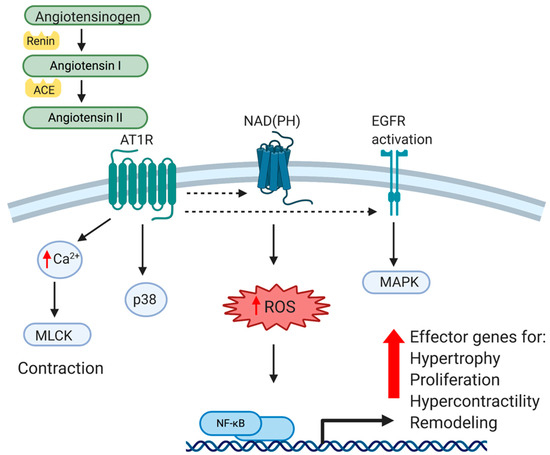
Figure 1. Cellular and Molecular effects of Angitions overlap and potentiate each other when we consider the many cellular and motensin II on VSMC. Angiotensin II (Ang II) is produced from the conversion of angiotensinogen to angiotensin I by renin, and then cleaved by angiotensin-converting enzyme (ACE) into Ang II. Ang II is recognized by a G-protein-coupled receptor, Angiotensin type 1 (AT1R). Canonical GPCR signaling activates receptor tyrosine kinases such as epidermal growth factor receptor (EGFR) as well as activating the NAD(P)H complex, resulting in generation of reactant oxygen species (ROS), a potent second messenger. In addition to regulation of VSMC contractility in a calcium/calmodulin-mediated pathway, AT1R stimulation results in activation of MAPK, and ultimately, NF-κB transactivation. Together, this leads to VSMC pathophysiological responses such as matrix production, hypertrophy, hypercontractility, vascular remodeling and hypertension.
3. Data, Model, Applications and Influences
3.1. Angiotensin II, Atherosclerosis, and Vascular Aging
Aging can be colnsidered a vascular similarities between them. These intersecting points are manifested in clinical studiedisease wherein decades of subclinical inflammatory and oxidative stress often results in arterial wall thickening, stiffening, and vulnerability to hypertension and atherosclerotic plaque development. Interestingly, the pro-inflammatory aging phenotype occurs with little to no mononuclear cell infiltrate, yet the vasculature is predisposed to inflammatory insult. Similarly, the incidence of clinically relevant hypertension increases exponentially with aging. Numerous cytokines such as platelet-derived growth factor (PDGF), matrix-inducing factors such as transforming growth factor-β, matrix proteins, matrix metalloproteinases, and vasoregulatory molecules such as endothelin (ET1) are increased in aged arteries, resulting in characteristic structural changes such as a thickened intima and a disorganized medial compartment [11]. Decades of in which lipisult from reactive oxygen species (ROS) lead to profound changes in the endothelium, including EC senescence and apoptosis [12][13]. In the media, lowering therapies reduce blohistologically, VSMC tend to be less organized, and elastic laminae less regular and fractured. Matrix proteins, particularly the collagens, are produced by and deposited around senescing VSMCs, and together with elastic fiber fragmentation reduce vascular compliance [14]. Arterial stiffening is associated pressure, and awith hypertension, atherosclerosis, and cerebrovascular incidents. In one study, right carotid artery stiffness was quantitated in old versus young individuals [15]. Young’s elasti-hypertensive medicc modulus (YEM), a quantitative measurement of material stiffness, was increased in the common carotid with advancing age, quantitatively determining that arterial stiffening accelerates with advanced age. Clearly, aging has deleterious effects on the vasculature similar to hypertension and atherosclerosis, but it is an unavoidable risk factor.
3.1.1. Aging and Angiotensin II
Expression of the aforementions reduce atherosclerotic plaque. At the med genes and ensuing structural and cellular changes are mediated and accelerated by the pro-inflammatory effects of Ang II. Ang II induces signal transduction events which mimic the aged arterial phenotype with respect to arterial remodeling events. In one example, the aortas of young rats infused with Ang II displayed a gene expression profile akin to older arteries with increased expression of TGFβ and TGFβ-responsive genes such as matrix and matrix degrading enzymes [16]. These aortas alecular leso displayed histological changes such as intimal and medial thickening similar to much older rats. Small arteries were isolated from human subjects and examined histologically and functionally by myography [17]. Structurally, arteries from hypertensive individual, the vass, regardless of age, resembled those of older individuals in terms of vascular smooth muscle cell (VSMC) is the target, integrator, and effremodeling, collagen deposition, and other fibrotic indices, all of which were compounded in aged hypertensive patients. Endothelium-dependent vasodilation was impaired in the hypertensive group and also correlated with the aged group. The authors also observed reduced nitric oxide availability in arteries from aged and hypertensive individuals, and concluded that oxidative stress in aged and hypertensive individuals was perhaps the driving force behind the increased vascular remodeling and reduced vascular reactivity. Furthermore, also in human subjects, the blockade of Ang II signaling not only reduced pro-inflammatory gene expression, but also delayed the development of age-associated aortic remodeling [18]. In one randomized clinical trial, elderly human subjects treated with the angiotensin receptor cell oblocker Valsartan demonstrated improved age-related vascular compliance [19]. In one longitudinal study, carotid artery stiffness was correlated with advancing age [20], but additioth atheron of anti-hypertensive therapy was significantly correlated with improved artery distensibility, linking age, artery stiffness, and hypertension.
At the cellular and molecular level, many of the age-associated chanic and the majorges in the vasculature associated with Ang II can be attributed to the many deleterious effector protein of the s of an increase in ROS. It is well accepted that oxidative stress can modify or otherwise irrevocably damage AND and other cellular machinery, indicative of many degenerative diseases, including hypertension, atherosclerosis, and especially ageing [21]. It has been shown in mice that disruption of AT1R prolongs longevity, which the signal investigators attributed to reduced ROS in the vascular wall [22]. These mice also developed reduced atherosclerosis as they aged. In other studies, Ang II-iotensin II (Angnduced ROS induced an aging phenotype in VSMC, as assayed by increased replicative senescence, increased DNA damage, and a reduction in p53-mediated telomere length II)[14]. Together, these studignals can potentiate each other and prime the es strongly suggest that the increased ROS induced by Ang II leads to enhanced VSMC biological aging. Consistent with the beneficial effects of AT2R engagement in terms of hypotension and attenuation of inflammatory signals, it was observed that over expression of the AT2R interacting protein ATIP, which regulates AT2R activity, reduced Ang II-induced VSMC senescence. Transgenic mice, which over express ATIP in aortic VSMC, demonstrated reduced senescence in thoracic aorta compared with control mice [23].
Similar tery and exacerbao that observed in VSMC, one study demonstrated that Ang II induced senescent changes in EC indicative of the aging phenotype, including decreased proliferation, increased apoptosis, and abnormal cell morphology [24]. Considering that EC dysfunction is one hypertension anof the earliest cellular harbingers of atherogenesis, this cellular event links Angiotensin II, aging, and atherosclerosis. Therefore, VSMCs are the fogether, aging appears to predispose vascular cells, particularly VSMC, to an inflammatory phenotype and plaque development, and subsequently sets the stage for the more aggressive pro-inflammatory events initiated by atherogenic events such as sub-endothelial deposition of oxidized LDL.
3.1.2. Aging and Atherosclerosis
An unavoidablcrum in progression of these diseases and, thereforee and unmodifiable risk factor for atherogenesis is advancing age. Expression of inflammatory cytokines, cell cycle proteins, and mitochondrial dysfunction have all been implicated as causative atherogenic events, and all are dysregulated in aged arteries. Experiments have been undertaken to determine if atherosclerosis can develop independently of chronic hyperlipidemia. At the systemic level, oxidative stress is increased with advancing age, as tissues from aged animals demonstrate increased ROS, which not only leads to increased oxidation of LDL, but age-related vascular remodeling similar to those described for Ang II-treated animals [25]. In one interesting study, it was founderstanding that increasing IL-6 levels correlated with mitochondrial dysfunction and mitophagy in aged versus young mice [26]. This study also she effects of aowed that atherogenesis increased in aged wild-type mice, and hyperlipidemia further increased not only atherogenesis, but IL-6 expression and mitophagy. This study was confirmed by another group who observed that IL-6 and adhesion molecules were significantly increased in aorta from aged versus young mice [27]. Together, these studies indicate that aged VSMC are predisposed to a pro-inflammatory phenotype, and thus vulnerable to atherogenic stimuli and Ang II on the. This also implicates inflammation and the pro-inflammatory cytokine IL-6 as a mediator of these effects. Another study in human atherosclerotic plaque determined that CAP VSMC is expressed senescent marker gene expression that was absent in control arteries [28]. These markers correlated with shortened telomeres, the length of which was inversely to understanproportional to the severity of atherosclerosis. The authors suggested that increased oxidative stress in proximity to the plaque drove the local VSMC into premature senescence.
4. Conclusions
The synthesis of the studies outlined in this review present an ing and treating atherosclerosis and hypertension.
2.timate relationship between hypertension, atherosclerosis, and aging. Chronic hypercholesterolemia induces the expression of many components of the RAS, and at multiple systemic and cellular targets, Ang II induces the expression of inflammatory and atherogenic proteins, which together leave the artery primed for hypertension and atherosclerosis. Inhibitors of Ang II synthesis and AT1R antagonists reduce vascular inflammation and plaque progression, and lipid-lowering therapy reduces blood pressure. In many murine studies, the genetic deletion of multiple components of the atherosclerotic pathway reduces hypertension, and deletion of members of the RAS Iattentroducuates atherogenesis. These studies are supported by clinical observation
2.1. Hypertension and Angiotensin II
3s in humans using pharmacological inhibitors. DBoth of these diseases ata, Model, Applire more prevent in aged arteries, and their severity is exacerbated by age. Morphologically, advanced hypertensive and atherosclerotic arteries resemble those from aged individuals. Atherogenic and hypertensive stimuli intersect on the VSMC suggesting that hypertension, atherosclerosis, and vascular smooth muscle cells make a perfect trio for vascular pathophysiology. The identifications and Influence of the molecular mediators of these diseases is key to our better understanding of them and, when supported by rigorous genetic studies in mice, represents an opportunity for the development of therapeutics to combat these vascular diseases which are approaching epic proportions
3.1. Angiotensin II, Atherosclerosis, and Vascular Aging
3.1.1. Aging and Angiotensin II
3.1.2. Aging and Atherosclerosis
.
4. Conclusions
The synthesis of the studies outlined in this review present an intimate relationship between hypertension, atherosclerosis, and aging. Chronic hypercholesterolemia induces the expression of many components of the RAS, and at multiple systemic and cellular targets, Ang II induces the expression of inflammatory and atherogenic proteins, which together leave the artery primed for hypertension and atherosclerosis. Inhibitors of Ang II synthesis and AT1R antagonists reduce vascular inflammation and plaque progression, and lipid-lowering therapy reduces blood pressure. In many murine studies, the genetic deletion of multiple components of the atherosclerotic pathway reduces hypertension, and deletion of members of the RAS attenuates atherogenesis. These studies are supported by clinical observations in humans using pharmacological inhibitors. Both of these diseases are more prevent in aged arteries, and their severity is exacerbated by age. Morphologically, advanced hypertensive and atherosclerotic arteries resemble those from aged individuals. Atherogenic and hypertensive stimuli intersect on the VSMC suggesting that hypertension, atherosclerosis, and vascular smooth muscle cells make a perfect trio for vascular pathophysiology. The identification of the molecular mediators of these diseases is key to our better understanding of them and, when supported by rigorous genetic studies in mice, represents an opportunity for the development of therapeutics to combat these vascular diseases which are approaching epic proportions.
References
- Wang, M.; Monticone, R.E.; Lakatta, E.G. Proinflammation of aging central arteries: A mini-review. Gerontology 2014, 60, 519–529. [Google Scholar] [CrossRef]
- Asai, K.; Kudej, R.K.; Shen, Y.T.; Yang, G.P.; Takagi, G.; Kudej, A.B.; Geng, Y.J.; Sato, N.; Nazareno, J.B.; Vatner, D.E.; et al. Peripheral vascular endothelial dysfunction and apoptosis in old monkeys. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1493–1499. [Google Scholar] [CrossRef]
- Brandes, R.P.; Fleming, I.; Busse, R. Endothelial aging. Cardiovasc. Res. 2005, 66, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Herbert, K.E.; Mistry, Y.; Hastings, R.; Poolman, T.; Niklason, L.; Williams, B. Angiotensin II-mediated oxidative DNA damage accelerates cellular senescence in cultured human vascular smooth muscle cells via telomere-dependent and independent pathways. Circ. Res. 2008, 102, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Gepner, A.D.; Korcarz, C.E.; Colangelo, L.A.; Hom, E.K.; Tattersall, M.C.; Astor, B.C.; Kaufman, J.D.; Liu, K.; Stein, J.H. Longitudinal effects of a decade of aging on carotid artery stiffness: The multiethnic study of atherosclerosis. Stroke 2014, 45, 48–53. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, J.; Spinetti, G.; Jiang, L.Q.; Monticone, R.; Zhao, D.; Cheng, L.; Krawczyk, M.; Talan, M.; Pintus, G.; et al. Angiotensin II activates matrix metalloproteinase type II and mimics age-associated carotid arterial remodeling in young rats. Am. J. Pathol. 2005, 167, 1429–1442. [Google Scholar] [CrossRef]
- Bruno, R.M.; Duranti, E.; Ippolito, C.; Segnani, C.; Bernardini, N.; Di Candio, G.; Chiarugi, M.; Taddei, S.; Virdis, A. Different Impact of Essential Hypertension on Structural and Functional Age-Related Vascular Changes. Hypertension 2017, 69, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Benigni, A.; Corna, D.; Zoja, C.; Sonzogni, A.; Latini, R.; Salio, M.; Conti, S.; Rottoli, D.; Longaretti, L.; Cassis, P.; et al. Disruption of the Ang II type 1 receptor promotes longevity in mice. J. Clin. Invest. 2009, 119, 524–530. [Google Scholar] [CrossRef]
- Min, L.J.; Mogi, M.; Iwanami, J.; Jing, F.; Tsukuda, K.; Ohshima, K.; Horiuchi, M. Angiotensin II type 2 receptor-interacting protein prevents vascular senescence. J. Am. Soc. Hypertens. 2012, 6, 179–184. [Google Scholar] [CrossRef]
- Shan, H.Y.; Bai, X.J.; Chen, X.M. Apoptosis is involved in the senescence of endothelial cells induced by angiotensin II. Cell Biol. Int. 2008, 32, 264–270. [Google Scholar] [CrossRef]
- Weiss, D.; Sorescu, D.; Taylor, W.R. Angiotensin II and atherosclerosis. Am. J. Cardiol. 2001, 87, 25–32. [Google Scholar] [CrossRef]
- Tyrrell, D.J.; Blin, M.G.; Song, J.; Wood, S.C.; Zhang, M.; Beard, D.A.; Goldstein, D.R. Age-Associated Mitochondrial Dysfunction Accelerates Atherogenesis. Circ. Res. 2020, 126, 298–314. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Shen, H.; Schenten, D.; Shan, P.; Lee, P.J.; Goldstein, D.R. Aging enhances the basal production of IL-6 and CCL2 in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Matthews, C.; Gorenne, I.; Scott, S.; Figg, N.; Kirkpatrick, P.; Ritchie, A.; Goddard, M.; Bennett, M. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: Effects of telomerase and oxidative stress. Circ. Res. 2006, 99, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Monticone, R.E.; Lakatta, E.G. Proinflammation of aging central arteries: A mini-review. Gerontology 2014, 60, 519–529. [Google Scholar] [CrossRef]
- Asai, K.; Kudej, R.K.; Shen, Y.T.; Yang, G.P.; Takagi, G.; Kudej, A.B.; Geng, Y.J.; Sato, N.; Nazareno, J.B.; Vatner, D.E.; et al. Peripheral vascular endothelial dysfunction and apoptosis in old monkeys. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1493–1499. [Google Scholar] [CrossRef]
- Brandes, R.P.; Fleming, I.; Busse, R. Endothelial aging. Cardiovasc. Res. 2005, 66, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Herbert, K.E.; Mistry, Y.; Hastings, R.; Poolman, T.; Niklason, L.; Williams, B. Angiotensin II-mediated oxidative DNA damage accelerates cellular senescence in cultured human vascular smooth muscle cells via telomere-dependent and independent pathways. Circ. Res. 2008, 102, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Gepner, A.D.; Korcarz, C.E.; Colangelo, L.A.; Hom, E.K.; Tattersall, M.C.; Astor, B.C.; Kaufman, J.D.; Liu, K.; Stein, J.H. Longitudinal effects of a decade of aging on carotid artery stiffness: The multiethnic study of atherosclerosis. Stroke 2014, 45, 48–53. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, J.; Spinetti, G.; Jiang, L.Q.; Monticone, R.; Zhao, D.; Cheng, L.; Krawczyk, M.; Talan, M.; Pintus, G.; et al. Angiotensin II activates matrix metalloproteinase type II and mimics age-associated carotid arterial remodeling in young rats. Am. J. Pathol. 2005, 167, 1429–1442. [Google Scholar] [CrossRef]
- Bruno, R.M.; Duranti, E.; Ippolito, C.; Segnani, C.; Bernardini, N.; Di Candio, G.; Chiarugi, M.; Taddei, S.; Virdis, A. Different Impact of Essential Hypertension on Structural and Functional Age-Related Vascular Changes. Hypertension 2017, 69, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Benigni, A.; Corna, D.; Zoja, C.; Sonzogni, A.; Latini, R.; Salio, M.; Conti, S.; Rottoli, D.; Longaretti, L.; Cassis, P.; et al. Disruption of the Ang II type 1 receptor promotes longevity in mice. J. Clin. Invest. 2009, 119, 524–530. [Google Scholar] [CrossRef]
- Min, L.J.; Mogi, M.; Iwanami, J.; Jing, F.; Tsukuda, K.; Ohshima, K.; Horiuchi, M. Angiotensin II type 2 receptor-interacting protein prevents vascular senescence. J. Am. Soc. Hypertens. 2012, 6, 179–184. [Google Scholar] [CrossRef]
- Shan, H.Y.; Bai, X.J.; Chen, X.M. Apoptosis is involved in the senescence of endothelial cells induced by angiotensin II. Cell Biol. Int. 2008, 32, 264–270. [Google Scholar] [CrossRef]
- Weiss, D.; Sorescu, D.; Taylor, W.R. Angiotensin II and atherosclerosis. Am. J. Cardiol. 2001, 87, 25–32. [Google Scholar] [CrossRef]
- Tyrrell, D.J.; Blin, M.G.; Song, J.; Wood, S.C.; Zhang, M.; Beard, D.A.; Goldstein, D.R. Age-Associated Mitochondrial Dysfunction Accelerates Atherogenesis. Circ. Res. 2020, 126, 298–314. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Shen, H.; Schenten, D.; Shan, P.; Lee, P.J.; Goldstein, D.R. Aging enhances the basal production of IL-6 and CCL2 in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Matthews, C.; Gorenne, I.; Scott, S.; Figg, N.; Kirkpatrick, P.; Ritchie, A.; Goddard, M.; Bennett, M. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: Effects of telomerase and oxidative stress. Circ. Res. 2006, 99, 156–164. [Google Scholar] [CrossRef] [PubMed]
References
- Kai M. Schmidt-Ott; Shuntaro Kagiyama; M. Ian Phillips; The multiple actions of angiotensin II in atherosclerosis. Regulatory Peptides 2000, 93, 65-77, 10.1016/s0167-0115(00)00178-6.
- R M Touyz; Gang He; Mohammed El Mabrouk; Quy Diep; Vartan Mardigyan; Ernesto L Schiffrin; Differential activation of extracellular signal-regulated protein kinase 1/2 and p38 mitogen activated-protein kinase by AT1 receptors in vascular smooth muscle cells from Wistar–Kyoto rats and spontaneously hypertensive rats. Journal of Hypertension 2001, 19, 553-559, 10.1097/00004872-200103001-00006.
- Lucchesi, P.A.; Bell, J.M.; Willis, L.S.; Byron, K.L.; Corson, M.A.; Berk, B.C; Ca(2+)-dependent mitogen-activated protein kinase activation in spontaneously hypertensive rat vascular smooth muscle defines a hypertensive signal transduction phenotype. Circ. Res. 1996, 78, 962–970.
- M Ruiz-Ortega; O Lorenzo; M Rupérez; Y Suzuki; J Egido; Angiotensin II activates nuclear transcription factor-kappaB in aorta of normal rats and in vascular smooth muscle cells of AT1 knockout mice. Nephrology Dialysis Transplantation 2001, 16, 27–33.
- A. Maziar Zafari; Masuko Ushio-Fukai; Marjorie Akers; Qiqin Yin; Aalok Shah; David G. Harrison; W. Robert Taylor; Kathy K Griendling; Role of NADH/NADPH Oxidase–Derived H2O2in Angiotensin II–Induced Vascular Hypertrophy. Hypertension 1998, 32, 488-495, 10.1161/01.hyp.32.3.488.
- Masuko Ushio-Fukai; A M Zafari; T Fukui; N Ishizaka; K K Griendling; p22phox is a critical component of the superoxide-generating NADH/NADPH oxidase system and regulates angiotensin II-induced hypertrophy in vascular smooth muscle cells.. Journal of Biological Chemistry 1996, 271, 23317–23321.
- K K Griendling; D Sorescu; Masuko Ushio-Fukai; NAD(P)H oxidase: role in cardiovascular biology and disease. Circulation Research 2000, 86, 494–501.
- G M Rubanyi; P M Vanhoutte; Superoxide anions and hyperoxia inactivate endothelium-derived relaxing factor. American Journal of Physiology-Legacy Content 1986, 250, H822–H827.
- M Ruiz-Ortega; O Lorenzo; M Rupérez; V Esteban; Y Suzuki; S Mezzano; J J Plaza; J Egido; Role of the renin-angiotensin system in vascular diseases: expanding the field. Hypertension 2001, 38, 1382–1387.
- B Schieffer; E Schieffer; D Hilfiker-Kleiner; A Hilfiker; P T Kovanen; M Kaartinen; J Nussberger; W Harringer; H. Drexler; Expression of angiotensin II and interleukin 6 in human coronary atherosclerotic plaques: potential implications for inflammation and plaque instability. Circulation 2000, 101, 1372–1378.
- Mingyi Wang; Robert E. Monticone; Edward G. Lakatta; Proinflammation of aging central arteries: a mini-review. Gerontology 2014, 60, 519-529, 10.1159/000362548.
- Kuniya Asai; Raymond K. Kudej; You-Tang Shen; Gui-Ping Yang; Gen Takagi; Amelia B. Kudej; Yong-Jian Geng; Naoki Sato; Jerome B. Nazareno; Rothy E. Vatner; et al.Filipinas NatividadSanford P. BishopStephen F. Vatner Peripheral vascular endothelial dysfunction and apoptosis in old monkeys. Arteriosclerosis, Thrombosis, and Vascular Biology 2000, 20, 1493-1499, 10.1161/01.atv.20.6.1493.
- Brandes, R.P.; Fleming, I.; Busse, R; Endothelial aging. Cardiovasc. Res. 2005, 66, 286–294.
- Karl Herbert; Yogita Mistry; Richard Hastings; Toryn Poolman; Laura Niklason; Bryan Williams; Angiotensin II-mediated oxidative DNA damage accelerates cellular senescence in cultured human vascular smooth muscle cells via telomere-dependent and independent pathways. Circulation Research 2007, 102, 201-208, 10.1161/CIRCRESAHA.107.158626.
- Adam D. Gepner; Claudia Korcarz; Laura A. Colangelo; Elizabeth K. Hom; Matthew C. Tattersall; Brad C. Astor; Joel D Kaufman; Kiang Liu; James H. Stein; Longitudinal effects of a decade of aging on carotid artery stiffness: the multiethnic study of atherosclerosis. Stroke 2014, 45, 48-53, 10.1161/STROKEAHA.113.002649.
- Mingyi Wang; Jing Zhang; Gaia Spinetti; Li-Qun Jiang; Robert Monticone; Di Zhao; Linda Cheng; Melissa Krawczyk; Mark Talan; Gianfranco Pintus; et al.Edward G. Lakatta Angiotensin II Activates Matrix Metalloproteinase Type II and Mimics Age-Associated Carotid Arterial Remodeling in Young Rats. The American Journal of Pathology 2005, 167, 1429-1442, 10.1016/s0002-9440(10)61229-1.
- Rosa Maria Bruno; Emiliano Duranti; Chiara Ippolito; Cristina Segnani; Nunzia Bernardini; Giulio Di Candio; Massimo Chiarugi; Stefano Taddei; Agostino Virdis; Different Impact of Essential Hypertension on Structural and Functional Age-Related Vascular Changes. Hypertension 2017, 69, 71-78, 10.1161/hypertensionaha.116.08041.
- Adam Oesterle; Ulrich Laufs; James K. Liao; Pleiotropic Effects of Statins on the Cardiovascular System. Circulation Research 2017, 120, 229-243, 10.1161/CIRCRESAHA.116.308537.
- Changping Hu; Abhijit Dandapat; Jiawei Chen; Yong Liu; Paul L. Hermonat; Robert M. Carey; Jawahar L. Mehta; Over-expression of angiotensin II type 2 receptor (agtr2) reduces atherogenesis and modulates LOX-1, endothelial nitric oxide synthase and heme-oxygenase-1 expression. Atherosclerosis 2008, 199, 288-294, 10.1016/j.atherosclerosis.2007.11.006.
- Seul-Gee Lee; Seung-Jun Lee; Nguyen Viet Phuong Thuy; Jung-Sun Kim; Jung-Jae Lee; Oh-Hyun Lee; Choong-Ki Kim; Jaewon Oh; Seil Park; Se Hoon Kim; et al.Sungha ParkSang-Hak LeeSung-Jin HongChul-Min AhnByeong-Keuk KimYoung-Guk KoDonghoon ChoiMyeong-Ki HongYangsoo JangOk-Hee Lee Synergistic protective effects of a statin and an angiotensin receptor blocker for initiation and progression of atherosclerosis. PLOS ONE 2019, 14, e0215604, 10.1371/journal.pone.0215604.
- Jurgen A. Marteijn; Hannes Lans; Wim Vermeulen; Jan H. J. Hoeijmakers; Understanding nucleotide excision repair and its roles in cancer and ageing. Nature Reviews Molecular Cell Biology 2014, 15, 465-481, 10.1038/nrm3822.
- Ariela Benigni; Daniela Corna; Carla Zoja; Aurelio Sonzogni; Roberto Latini; Monica Salio; Sara Conti; Daniela Rottoli; Lorena Longaretti; Paola Cassis; et al.Marina MorigiThomas M. CoffmanGiuseppe Remuzzi Disruption of the Ang II type 1 receptor promotes longevity in mice. Journal of Clinical Investigation 2009, 119, 524-530, 10.1172/JCI36703.
- Li-Juan Min; Masaki Mogi; Jun Iwanami; Fei Jing; Kana Tsukuda; Kousei Ohshima; Masatsugu Horiuchi; Angiotensin II type 2 receptor-interacting protein prevents vascular senescence. Journal of the American Society of Hypertension 2012, 6, 179-184, 10.1016/j.jash.2012.01.006.
- Hai‐Yan Shan; Xiao‐Juan Bai; Xiang‐Mei Chen; Apoptosis is involved in the senescence of endothelial cells induced by angiotensin II. Cell Biology International 2008, 32, 264-270, 10.1016/j.cellbi.2007.09.003.
- Weiss, D.; Sorescu, D.; Taylor, W.R; Angiotensin II and atherosclerosis. Am. J. Cardiol. 2001, 87, 25–32.
- Daniel J. Tyrrell; Muriel G. Blin; Jianrui Song; Sherri C. Wood; Min Zhang; Daniel A. Beard; Daniel R. Goldstein; Age-Associated Mitochondrial Dysfunction Accelerates Atherogenesis. Circulation Research 2020, 126, 298-314, 10.1161/circresaha.119.315644.
- Song, Y.; Shen, H.; Schenten, D.; Shan, P.; Lee, P.J.; Goldstein, D.R; Aging enhances the basal production of IL-6 and CCL2 in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 103–109.
- Matthews, C.; Gorenne, I.; Scott, S.; Figg, N.; Kirkpatrick, P.; Ritchie, A.; Goddard, M.; Bennett, M; Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: Effects of telomerase and oxidative stress. Circ. Res. 2006, 99, 156–164.